Single-cell copy-number variation with inferCNV#
This tutorial walks through ov.single.CNV(method='infercnv') — a
wrapper around the pure-Python
py-inferCNV re-implementation
of the Broad Institute’s R inferCNV
(Tickle et al., 2019).
inferCNV is reference-anchored: you tell it which cell-type categories to treat as diploid normals, and the resulting CN matrix is each cell’s deviation from that reference. This is more sensitive than CopyKAT when a clean normal population exists in the same dataset (e.g. immune cells in a tumour biopsy), at the cost of needing a cell-type annotation.
Why use
omicverse/py-inferCNVinstead oficbi-lab/infercnvpy?
infercnvpy(ICBI Lab) is a lightweight Python wrapper that re-implements R inferCNV’s Phase 1 (preprocessing) pipeline only. It explicitly omits Phases 2 and 3 — the HMM-based CNV region calling and Bayesian filtering steps that are central to R inferCNV’s scientific output. This is acknowledged by the maintainer in #1 (“HMM-based CNV caller”, open since 2021) and #121 (“stable release”, closed 2024 with “I don’t have the time to further pursue this project”). The repo has accumulated 18 open issues including long-standing gaps on gene-level CNV extraction (#50, open since 2022) and memory scaling (#180, 2026). In contrast,py-inferCNVships Phase 2 (HMM i3/i6 state calling, tumour subclustering) and Phase 3 (BayesNet filtering, denoising) as of v0.2.0, with bit-exact R-parity validation on Phase 1 (max diff < 1e-10) and Spearman ρ = 1.0000 on Phase 2 across multiple 10x patients — see itsNAMESPACE_PARITY.mdfor the audit table. The omicverse wrapper consumes it directly, no R needed.
Part.1 The math behind inferCNV#
inferCNV’s Phase 1 pipeline:
1. Filter + normalise. Low-expression genes are dropped; counts are normalised by sequencing depth and log-transformed $\(\;\;\tilde x_{ij} = \log_2(c_{ij} + 1)\)\( where \)c_{ij}\( is the depth-normalised count of gene \)j\( in cell \)i$.
2. Reference subtraction. For each gene, the mean expression of reference cells is subtracted from every cell: $\(\;\;r_{ij} = \tilde x_{ij} - \overline{\tilde x_{j,\,\text{ref}}}.\)$
3. Pyramidal smoothing. A weighted moving average of width window_length
(default 101) is applied along each chromosome to suppress per-gene noise
while preserving copy-number breakpoints.
4. Centre cells. The median of each cell’s smoothed profile is subtracted so the diploid baseline sits at zero.
5. Second reference subtraction + log inversion. Repeats step 2 on the smoothed values, then maps log-ratios back through \(2^x - 1\) to yield a per-bin CN deviation.
6. Outlier pruning. Bins exceeding lfc_clip (default 3) are clipped
to suppress experimental artefacts.
Reference: Patel et al., Science (2014) — original method; broadinstitute/inferCNV — R implementation.
Part.2 Load data#
Maynard et al. (2020) lung adenocarcinoma, 3 000 cells. The file already
carries gene-coordinate metadata in adata.var (chromosome, start,
end) — inferCNV uses these to order genes along the genome.
import omicverse as ov
ov.plot_set()
🔬 Starting plot initialization...
🧬 Detecting GPU devices…
✅ NVIDIA CUDA GPUs detected: 1
• [CUDA 0] NVIDIA H100 80GB HBM3
Memory: 79.1 GB | Compute: 9.0
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.1.3rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
import os
import scanpy as sc
import omicverse as ov
# Maynard et al. (2020) lung adenocarcinoma scRNA-seq — 3 000 cells, 55 556
# genes, gene coordinates already in adata.var. Bundled by infercnvpy.
DATA_URL = "https://github.com/icbi-lab/infercnvpy/releases/download/d0.1.0/maynard2020_3k.h5ad"
data_dir = "data/cnv"
os.makedirs(data_dir, exist_ok=True)
adata = sc.read(f"{data_dir}/maynard2020_3k.h5ad", backup_url=DATA_URL)
adata
AnnData object with n_obs × n_vars = 3000 × 55556
obs: 'age', 'sex', 'sample', 'patient', 'cell_type'
var: 'ensg', 'mito', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_counts', 'chromosome', 'start', 'end', 'gene_id', 'gene_name'
uns: 'cell_type_colors', 'neighbors', 'umap'
obsm: 'X_scVI', 'X_umap'
obsp: 'connectivities', 'distances'
Part.3 Identify reference cells#
Reference cells must be biologically diploid. In a tumour biopsy the infiltrating immune cells (T cells, B cells, macrophages, monocytes, NK cells) are the canonical choice — they’re abundant, homogeneous, and near-certain to have a normal karyotype. Stromal cells (fibroblasts, endothelial) can also be used.
Here we use the four largest non-epithelial categories as references. Epithelial cells become the query (potentially aneuploid) population.
reference_cats = ['T cell CD4', 'T cell CD8', 'Macrophage', 'B cell']
print('Reference cells (diploid):',
int(adata.obs["cell_type"].isin(reference_cats).sum()))
print('Query cells (epithelial / other):',
int(adata.obs["cell_type"].isin(['Epithelial cell']).sum()),
'(Epithelial cell only)')
Reference cells (diploid): 997
Query cells (epithelial / other): 522 (Epithelial cell only)
Part.4 Run inferCNV#
ov.single.CNV(method='infercnv') writes the same unified schema as
CopyKAT — same plot calls work unchanged.
slot |
content |
|---|---|
|
cells × ~4 k bins of log-CN |
|
chromosome → starting-bin index |
|
per-cell mean abs(log-CN) — proxy for “tumour-ness” |
inferCNV does not classify cells by default — adata.obs['cnv_prediction']
stays NaN. To threshold cnv_score into tumour/normal after the fact, see
the bottom of the notebook.
cnv = ov.single.CNV(adata, method='infercnv')
cnv.run(reference_key='cell_type', reference_cat=reference_cats,
exclude_chromosomes=('chrX', 'chrY', 'chrM'), verbose=False)
<omicverse.single._cnv.CNV at 0x7f14550906a0>
adata.obs[['cnv_score']].describe().T
| count | mean | std | min | 25% | 50% | 75% | max | |
|---|---|---|---|---|---|---|---|---|
| cnv_score | 3000.0 | 0.073123 | 0.017058 | 0.028157 | 0.061502 | 0.072854 | 0.08243 | 0.233666 |
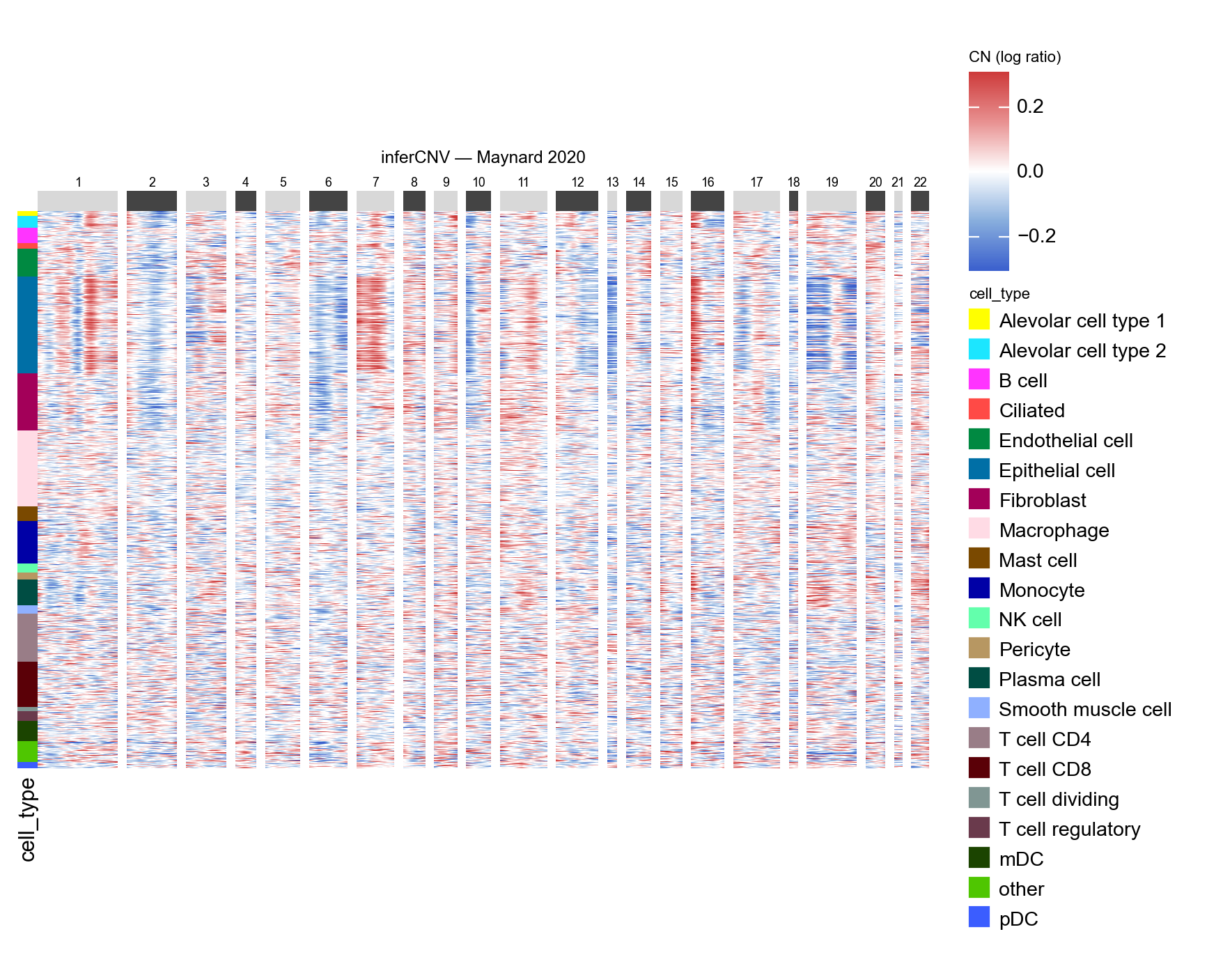
Part.5 Genome-wide heatmap#
Plotting the cells grouped by their original cell_type annotation lets
us see whether tumour-like CN patterns concentrate in the Epithelial-cell
band. Immune and stromal cells should sit near zero (they were the
reference); Epithelial cells should show recurrent gains and losses.
groupby picks a single adata.obs column for row ordering. If you
want an extra colour strip on top without changing the order, pass it
via annotations — see the CopyKAT tutorial for an example.
ov.pl.cnv_heatmap(adata, groupby='cell_type',
figsize=(8, 5), title='inferCNV — Maynard 2020')
<marsilea.heatmap.Heatmap at 0x7f14550a3e80>
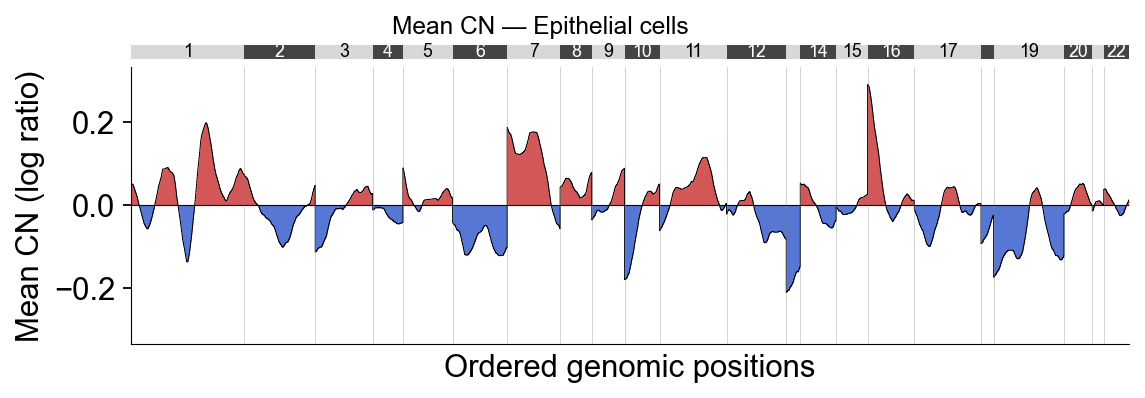
Part.6 Mean CN summary#
Mean CN per bin restricted to Epithelial cells — the pseudo-bulk tumour profile.
ov.pl.cnv_summary(adata, groupby='cell_type', subset='Epithelial cell',
figsize=(8, 2.6), title='Mean CN — Epithelial cells')
(<Figure size 640x208 with 2 Axes>,
{'line': <Axes: xlabel='Ordered genomic positions', ylabel='Mean CN (log ratio)'>,
'ideogram': <Axes: >})
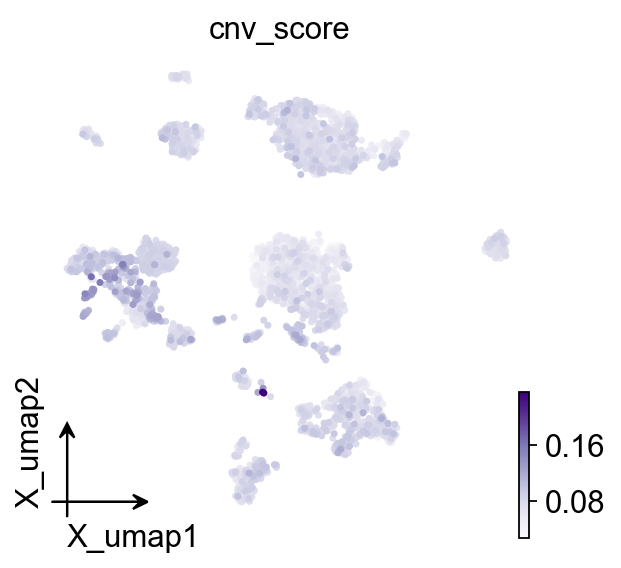
Part.7 UMAP#
inferCNV doesn’t classify cells, but cnv_score (mean absolute CN
deviation) is a useful continuous “tumour-ness” gradient. Overlaying it
on the existing UMAP shows whether the score peaks at the Epithelial
cluster.
ov.pl.cnv_umap(adata, color='cnv_score')
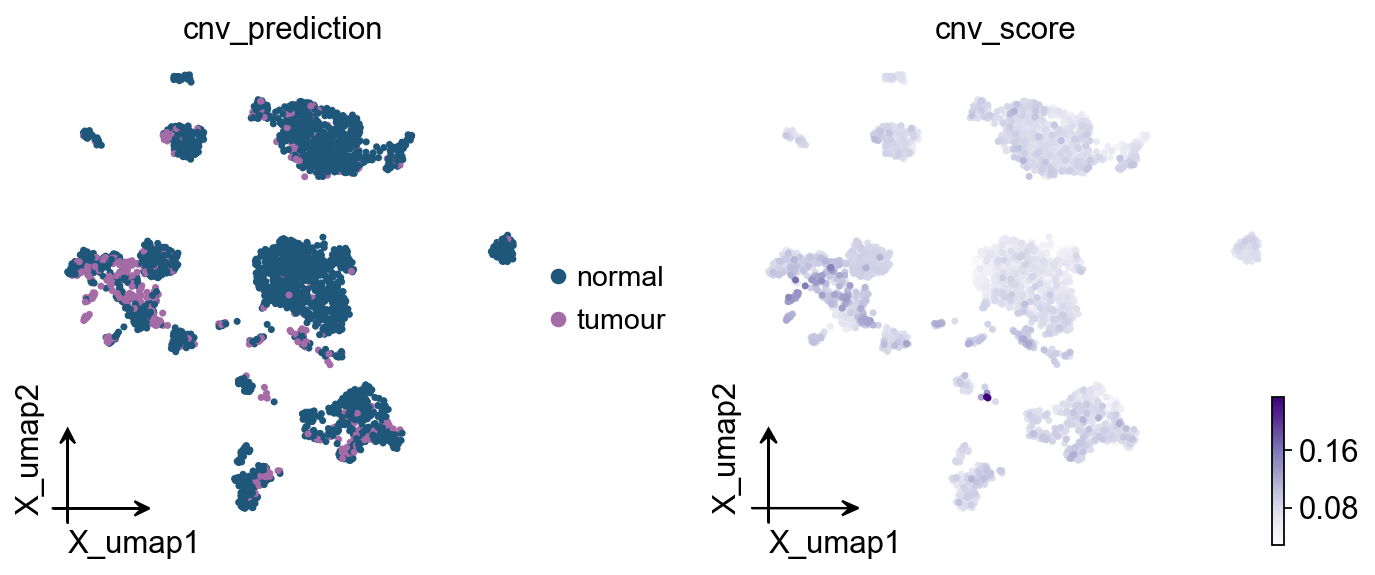
Thresholding cnv_score into a tumour / normal call#
If you want a categorical call on top of inferCNV, a simple threshold on
cnv_score (e.g. > 90th percentile of reference cells) works well:
import numpy as np
ref_mask = adata.obs['cell_type'].isin(reference_cats).values
ref_threshold = np.percentile(adata.obs.loc[ref_mask, 'cnv_score'], 95)
adata.obs['cnv_prediction'] = np.where(
adata.obs['cnv_score'] > ref_threshold, 'tumour', 'normal'
)
adata.obs['cnv_prediction'] = adata.obs['cnv_prediction'].astype('category')
adata.obs['cnv_prediction'].value_counts()
cnv_prediction
normal 2544
tumour 456
Name: count, dtype: int64
ov.pl.cnv_umap(adata, color=['cnv_prediction', 'cnv_score'])
Next steps: for unsupervised CN inference (no reference annotation
needed), see the CopyKAT tutorial. The two methods
can disagree at the margins — running both on the same dataset and
comparing their cnv_score columns side-by-side
(ov.pl.embedding(adata, color=['cnv_score_copykat', 'cnv_score_infercnv']))
is a useful sanity check.