Recommended workflow: SEACells end-to-end + downstream sanity#
This is the default tutorial for users new to ov.single.MetaCell. We
run the recommended backend ('seacells') on a typical single-sample
dataset and immediately drive it into the two most common downstream
analyses — differential expression and marker-dotplot — to show that the
metacell-level AnnData is a drop-in replacement for the cell-level one.
After this notebook:
Run
t_metacell_multisampleif you have ≥2 samples / batches.Browse zoo/index if you want to swap out the backend (faster:
kmeans/supercell; out-of-sample:metaq; sanity floor:random).
1. Setup#
import warnings
warnings.filterwarnings('ignore')
import numpy as np
import pandas as pd
import omicverse as ov
import scvelo as scv # demo dataset only
ov.plot_set()
🔬 Starting plot initialization...
🧬 Detecting GPU devices…
✅ NVIDIA CUDA GPUs detected: 1
• [CUDA 0] NVIDIA H100 80GB HBM3
Memory: 79.1 GB | Compute: 9.0
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.2.0 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
2. Load and preprocess#
Standard omicverse flow: qc → preprocess → scale → pca → neighbors → umap.
SEACells builds its kernel on a low-dim embedding (here X_pca).
adata = scv.datasets.pancreas()
adata = ov.pp.qc(adata,
tresh={'mito_perc': 0.20, 'nUMIs': 500, 'detected_genes': 250},
mt_startswith='mt-')
adata = ov.pp.preprocess(adata, mode='shiftlog|pearson', n_HVGs=2000)
adata.layers['lognorm'] = adata.X.copy()
adata = adata[:, adata.var.highly_variable_features]
ov.pp.scale(adata)
ov.pp.pca(adata, layer='scaled', n_pcs=30)
adata.obsm['X_pca'] = adata.obsm['scaled|original|X_pca']
ov.pp.neighbors(adata, n_neighbors=15, use_rep='X_pca')
ov.pp.umap(adata)
print('adata:', adata.shape, 'celltypes:', sorted(adata.obs['clusters'].unique()))
🖥️ Using CPU mode for QC...
📊 Step 1: Calculating QC Metrics
✓ Gene Family Detection:
┌──────────────────────────────┬────────────────────┬────────────────────┐
│ Gene Family │ Genes Found │ Detection Method │
├──────────────────────────────┼────────────────────┼────────────────────┤
│ Mitochondrial │ 13 │ Auto (MT-) │
├──────────────────────────────┼────────────────────┼────────────────────┤
│ Ribosomal │ 0 ⚠️ │ Auto (RPS/RPL) │
├──────────────────────────────┼────────────────────┼────────────────────┤
│ Hemoglobin │ 0 ⚠️ │ Auto (regex) │
└──────────────────────────────┴────────────────────┴────────────────────┘
✓ QC Metrics Summary:
┌─────────────────────────┬────────────────────┬─────────────────────────┐
│ Metric │ Mean │ Range (Min - Max) │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ nUMIs │ 6675 │ 3020 - 18524 │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ Detected Genes │ 2516 │ 1473 - 4492 │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ Mitochondrial % │ 0.7% │ 0.2% - 4.3% │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ Ribosomal % │ 0.0% │ 0.0% - 0.0% │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ Hemoglobin % │ 0.0% │ 0.0% - 0.0% │
└─────────────────────────┴────────────────────┴─────────────────────────┘
📈 Original cell count: 3,696
🔧 Step 2: Quality Filtering (SEURAT)
Thresholds: mito≤0.2, nUMIs≥500, genes≥250
📊 Seurat Filter Results:
• nUMIs filter (≥500): 0 cells failed (0.0%)
• Genes filter (≥250): 0 cells failed (0.0%)
• Mitochondrial filter (≤0.2): 0 cells failed (0.0%)
✓ Filters applied successfully
✓ Combined QC filters: 0 cells removed (0.0%)
🎯 Step 3: Final Filtering
Parameters: min_genes=200, min_cells=3
Ratios: max_genes_ratio=1, max_cells_ratio=1
✓ Final filtering: 0 cells, 12,261 genes removed
🔍 Step 4: Doublet Detection
💡 Running pyscdblfinder (Python port of R scDblFinder)
🔍 Running scdblfinder detection...
[ScDblFinder] wrote scDblFinder_score + scDblFinder_class — threshold=0.387
✓ scDblFinder completed: 66 doublets removed (1.8%)
╭─ SUMMARY: qc ──────────────────────────────────────────────────────╮
│ Duration: 18.8586s │
│ Shape: 3,696 x 27,998 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● OBS │ ✚ cell_complexity (float) │
│ │ ✚ detected_genes (int) │
│ │ ✚ hb_perc (float) │
│ │ ✚ mito_perc (float) │
│ │ ✚ nUMIs (float) │
│ │ ✚ n_counts (float) │
│ │ ✚ n_genes (int) │
│ │ ✚ n_genes_by_counts (int) │
│ │ ✚ passing_mt (bool) │
│ │ ✚ passing_nUMIs (bool) │
│ │ ✚ passing_ngenes (bool) │
│ │ ✚ pct_counts_hb (float) │
│ │ ✚ pct_counts_mt (float) │
│ │ ✚ pct_counts_ribo (float) │
│ │ ✚ ribo_perc (float) │
│ │ ✚ total_counts (float) │
│ │
│ ● VAR │ ✚ hb (bool) │
│ │ ✚ mt (bool) │
│ │ ✚ ribo (bool) │
│ │
╰────────────────────────────────────────────────────────────────────╯
🔍 [2026-05-19 18:44:27] Running preprocessing in 'cpu' mode...
Begin robust gene identification
After filtration, 15737/15737 genes are kept.
Among 15737 genes, 15736 genes are robust.
✅ Robust gene identification completed successfully.
Begin size normalization: shiftlog and HVGs selection pearson
🔍 Count Normalization:
Target sum: 500000.0
Exclude highly expressed: True
Max fraction threshold: 0.2
⚠️ Excluding 1 highly-expressed genes from normalization computation
Excluded genes: ['Ghrl']
✅ Count Normalization Completed Successfully!
✓ Processed: 3,630 cells × 15,736 genes
✓ Runtime: 0.24s
🔍 Highly Variable Genes Selection (Experimental):
Method: pearson_residuals
Target genes: 2,000
Theta (overdispersion): 100
✅ Experimental HVG Selection Completed Successfully!
✓ Selected: 2,000 highly variable genes out of 15,736 total (12.7%)
✓ Results added to AnnData object:
• 'highly_variable': Boolean vector (adata.var)
• 'highly_variable_rank': Float vector (adata.var)
• 'highly_variable_nbatches': Int vector (adata.var)
• 'highly_variable_intersection': Boolean vector (adata.var)
• 'means': Float vector (adata.var)
• 'variances': Float vector (adata.var)
• 'residual_variances': Float vector (adata.var)
Time to analyze data in cpu: 1.48 seconds.
✅ Preprocessing completed successfully.
Added:
'highly_variable_features', boolean vector (adata.var)
'means', float vector (adata.var)
'variances', float vector (adata.var)
'residual_variances', float vector (adata.var)
'counts', raw counts layer (adata.layers)
End of size normalization: shiftlog and HVGs selection pearson
╭─ SUMMARY: preprocess ──────────────────────────────────────────────╮
│ Duration: 1.8644s │
│ Shape: 3,630 x 15,737 -> 3,630 x 15,736 │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● VAR │ ✚ highly_variable (bool) │
│ │ ✚ highly_variable_features (bool) │
│ │ ✚ highly_variable_rank (float) │
│ │ ✚ means (float) │
│ │ ✚ n_cells (int) │
│ │ ✚ percent_cells (float) │
│ │ ✚ residual_variances (float) │
│ │ ✚ robust (bool) │
│ │ ✚ variances (float) │
│ │
│ ● UNS │ ✚ history_log │
│ │ ✚ hvg │
│ │ ✚ log1p │
│ │
│ ● LAYERS │ ✚ counts (sparse matrix, 3630x15736) │
│ │
╰────────────────────────────────────────────────────────────────────╯
╭─ SUMMARY: scale ───────────────────────────────────────────────────╮
│ Duration: 0.6108s │
│ Shape: 3,630 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● LAYERS │ ✚ scaled (array, 3630x2000) │
│ │
╰────────────────────────────────────────────────────────────────────╯
computing PCA🔍
with n_comps=30
🖥️ Using sklearn PCA for CPU computation
🖥️ sklearn PCA backend: CPU computation
📊 PCA input data type: ArrayView, shape: (3630, 2000), dtype: float64
🔧 PCA solver used: covariance_eigh
finished✅ (2.21s)
╭─ SUMMARY: pca ─────────────────────────────────────────────────────╮
│ Duration: 2.2184s │
│ Shape: 3,630 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ scaled|original|cum_sum_eigenvalues │
│ │ ✚ scaled|original|pca_var_ratios │
│ │
│ ● OBSM │ ✚ scaled|original|X_pca (array, 3630x30) │
│ │
╰────────────────────────────────────────────────────────────────────╯
🖥️ Using Scanpy CPU to calculate neighbors...
🔍 K-Nearest Neighbors Graph Construction:
Mode: cpu
Neighbors: 15
Method: umap
Metric: euclidean
Representation: X_pca
🔍 Computing neighbor distances...
🔍 Computing connectivity matrix...
💡 Using UMAP-style connectivity
✓ Graph is fully connected
✅ KNN Graph Construction Completed Successfully!
✓ Processed: 3,630 cells with 15 neighbors each
✓ Results added to AnnData object:
• 'neighbors': Neighbors metadata (adata.uns)
• 'distances': Distance matrix (adata.obsp)
• 'connectivities': Connectivity matrix (adata.obsp)
╭─ SUMMARY: neighbors ───────────────────────────────────────────────╮
│ Duration: 8.4673s │
│ Shape: 3,630 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
╰────────────────────────────────────────────────────────────────────╯
🔍 [2026-05-19 18:44:41] Running UMAP in 'cpu' mode...
🖥️ Using Scanpy CPU UMAP...
🔍 UMAP Dimensionality Reduction:
Mode: cpu
Method: umap
Components: 2
Min distance: 0.5
{'n_neighbors': 15, 'method': 'umap', 'random_state': 0, 'metric': 'euclidean', 'use_rep': 'X_pca'}
🔍 Computing UMAP parameters...
🔍 Computing UMAP embedding (classic method)...
✅ UMAP Dimensionality Reduction Completed Successfully!
✓ Embedding shape: 3,630 cells × 2 dimensions
✓ Results added to AnnData object:
• 'X_umap': UMAP coordinates (adata.obsm)
• 'umap': UMAP parameters (adata.uns)
✅ UMAP completed successfully.
╭─ SUMMARY: umap ────────────────────────────────────────────────────╮
│ Duration: 0.8242s │
│ Shape: 3,630 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ umap │
│ │ └─ params: {'a': 0.5830300199950147, 'b': 1.334166993228519}│
│ │
╰────────────────────────────────────────────────────────────────────╯
adata: (3630, 2000) celltypes: ['Alpha', 'Beta', 'Delta', 'Ductal', 'Epsilon', 'Ngn3 high EP', 'Ngn3 low EP', 'Pre-endocrine']
3. Fit SEACells#
n_metacells = adata.n_obs // 50 is a reasonable starting point — it gives
~70–80 metacells per 4 k cells, with mean metacell size ~50 cells.
mc = ov.single.MetaCell(
adata.copy(), method='seacells',
n_metacells=adata.n_obs // 50,
use_rep='X_pca', device='cpu', random_state=0,
).fit()
print(f'fit done: n_metacells={mc.n_metacells}, '
f'runtime={mc._fit_result.runtime_s:.2f} s, '
f'capabilities={sorted(mc.capabilities)}')
Welcome to SEACells!
Parameter graph_construction = union being used to build KNN graph...
Building kernel on X_pca
fit done: n_metacells=72, runtime=11.64 s, capabilities=['latent', 'soft']
4. Aggregate to a metacell AnnData#
# 'sum' aggregation preserves raw-count totals — required by SCENIC / pseudobulk
# DE / CellPhoneDB. Use 'mean' for visualization-only workflows.
ad_mc = mc.predicted(method='soft', layer='counts', summary='sum',
celltype_label='clusters')
print(f'metacell AnnData: {ad_mc.shape}')
print(f' mean cells/metacell: {ad_mc.obs["n_cells"].mean():.1f}')
print(f' mean purity : {ad_mc.obs["clusters_purity"].mean():.3f}')
ad_mc.obs.head()
metacell AnnData: (72, 2000)
mean cells/metacell: 111.4
mean purity : 0.877
| n_cells | clusters | clusters_purity | |
|---|---|---|---|
| mc-0 | 200 | Alpha | 0.979167 |
| mc-1 | 45 | Epsilon | 1.000000 |
| mc-2 | 152 | Beta | 1.000000 |
| mc-3 | 112 | Beta | 1.000000 |
| mc-4 | 92 | Ductal | 0.975000 |
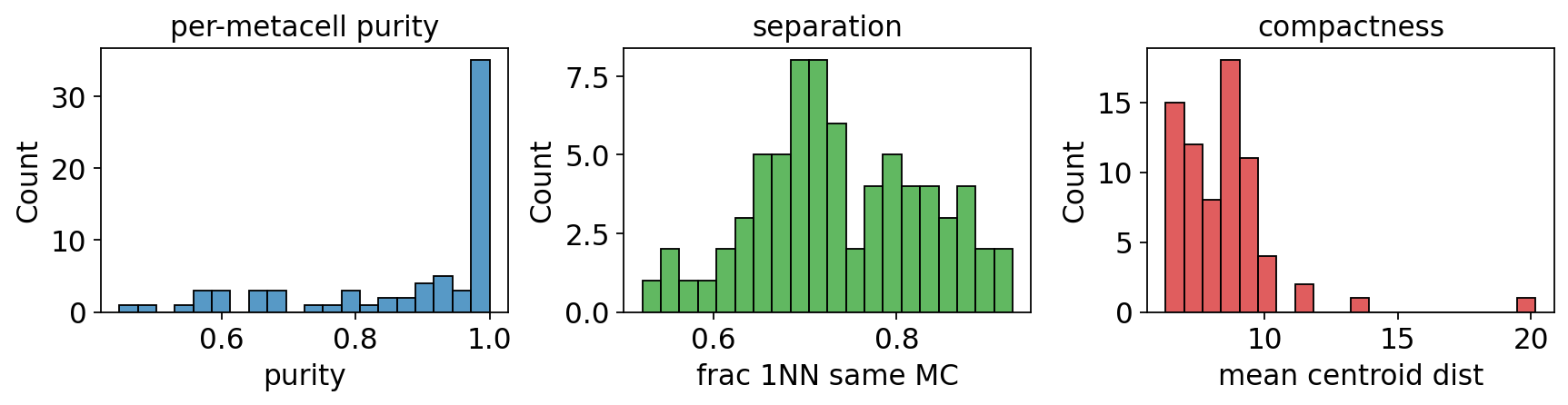
5. Quality check: purity / separation / compactness#
These three SEACells-style metrics apply to any metacell partition. All three are computed in one helper call and the histograms tell you whether the partition is honest.
purity, separation, compactness = ov.pl.metacell_metrics(
mc, label_key='clusters', use_rep='X_pca',
)
6. mcRigor: statistical validation#
Asks per metacell: is its gene–gene covariance larger than expected from a
within-cell gene-shuffle null at this metacell size? Metacells whose
mcDiv exceeds the size-stratified threshold are flagged as 'dubious'.
Lower dubious_rate → tighter metacells.
rep = mc.check_rigor(layer_lognorm='lognorm', n_rep=30,
feature_use=1000, random_state=0)
print(f'rigor_score : {rep.score:.3f}')
print(f'dubious_rate: {rep.dubious_rate:.3f}')
print(f'zero_rate : {rep.zero_rate:.3f}')
rigor_score : 0.555
dubious_rate: 0.647
zero_rate : 0.243
ov.pl.rigor_scatter(rep)
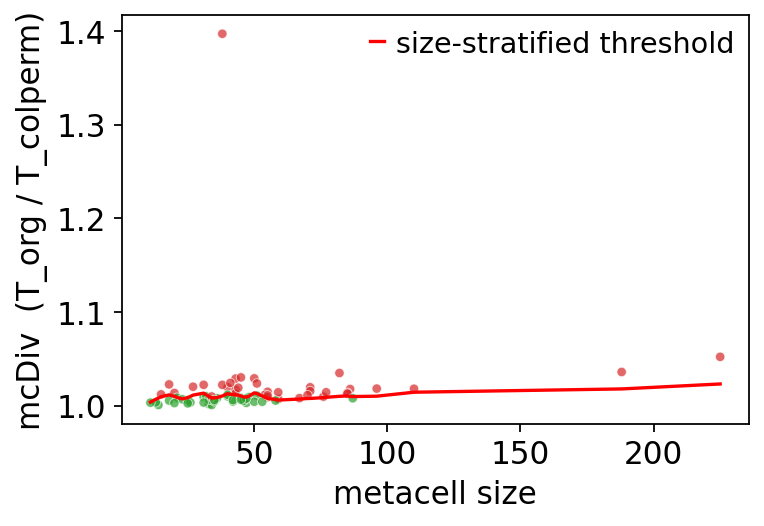
<Axes: xlabel='metacell size', ylabel='mcDiv (T_org / T_colperm)'>
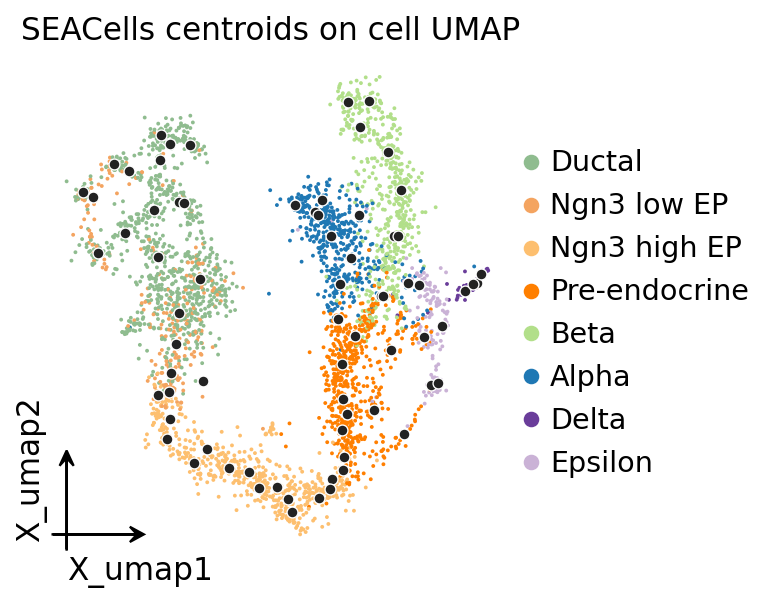
7. Visualize: metacell centroids on the source UMAP#
Centroids inside clearly-coloured cell-type islands = good metacells. Centroids straddling cell-type boundaries → mixed metacells (high mcDiv, low purity).
import matplotlib.pyplot as plt
fig, ax = plt.subplots(figsize=(5, 4))
ov.pl.embedding(mc.adata, basis='X_umap', color='clusters', ax=ax, show=False,
frameon='small', title='SEACells centroids on cell UMAP', size=12)
labels = mc._fit_result.assignments
pts = np.array([mc.adata.obsm['X_umap'][labels == u].mean(axis=0)
for u in np.unique(labels)])
ax.scatter(pts[:, 0], pts[:, 1], s=24, c='#222',
edgecolors='white', linewidths=0.6, zorder=5)
plt.tight_layout(); plt.show()
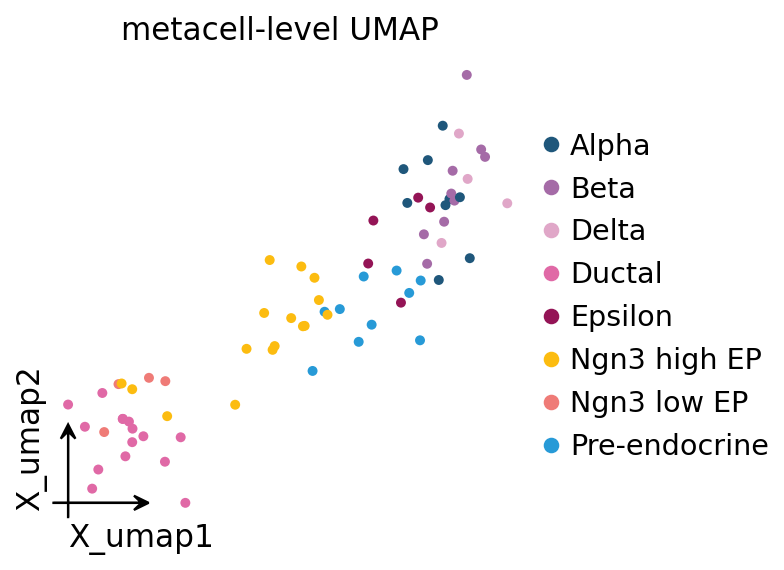
8. Visualize: metacell-level UMAP#
A common downstream use of metacells is to treat them as a much smaller atlas — re-run the standard preprocess → PCA → UMAP loop on the aggregated AnnData. Celltype structure should survive cleanly.
ad_mc = ov.pp.preprocess(ad_mc, mode='shiftlog|pearson',
n_HVGs=min(2000, ad_mc.n_vars))
ad_mc = ad_mc[:, ad_mc.var.highly_variable_features]
ov.pp.scale(ad_mc)
ov.pp.pca(ad_mc, layer='scaled', n_pcs=min(30, ad_mc.n_obs - 1))
ad_mc.obsm['X_pca'] = ad_mc.obsm['scaled|original|X_pca']
ov.pp.neighbors(ad_mc, n_neighbors=min(15, ad_mc.n_obs - 1), use_rep='X_pca')
ov.pp.umap(ad_mc)
ov.pl.embedding(ad_mc, basis='X_umap', color='clusters',
frameon='small', title='metacell-level UMAP', size=80)
🔍 [2026-05-19 18:45:28] Running preprocessing in 'cpu' mode...
Begin robust gene identification
After filtration, 2000/2000 genes are kept.
Among 2000 genes, 2000 genes are robust.
✅ Robust gene identification completed successfully.
Begin size normalization: shiftlog and HVGs selection pearson
🔍 Count Normalization:
Target sum: 500000.0
Exclude highly expressed: True
Max fraction threshold: 0.2
⚠️ Excluding 1 highly-expressed genes from normalization computation
Excluded genes: ['Ghrl']
✅ Count Normalization Completed Successfully!
✓ Processed: 72 cells × 2,000 genes
✓ Runtime: 0.00s
🔍 Highly Variable Genes Selection (Experimental):
Method: pearson_residuals
Target genes: 2,000
Theta (overdispersion): 100
✅ Experimental HVG Selection Completed Successfully!
✓ Selected: 2,000 highly variable genes out of 2,000 total (100.0%)
✓ Results added to AnnData object:
• 'highly_variable': Boolean vector (adata.var)
• 'highly_variable_rank': Float vector (adata.var)
• 'highly_variable_nbatches': Int vector (adata.var)
• 'highly_variable_intersection': Boolean vector (adata.var)
• 'means': Float vector (adata.var)
• 'variances': Float vector (adata.var)
• 'residual_variances': Float vector (adata.var)
Time to analyze data in cpu: 0.03 seconds.
✅ Preprocessing completed successfully.
Added:
'highly_variable_features', boolean vector (adata.var)
'means', float vector (adata.var)
'variances', float vector (adata.var)
'residual_variances', float vector (adata.var)
'counts', raw counts layer (adata.layers)
End of size normalization: shiftlog and HVGs selection pearson
╭─ SUMMARY: preprocess ──────────────────────────────────────────────╮
│ Duration: 0.0383s │
│ Shape: 72 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ REFERENCE_MANU │
│ │ ✚ _ov_provenance │
│ │ ✚ history_log │
│ │ ✚ hvg │
│ │ ✚ log1p │
│ │ ✚ status │
│ │ ✚ status_args │
│ │
│ ● LAYERS │ ✚ counts (sparse matrix, 72x2000) │
│ │
╰────────────────────────────────────────────────────────────────────╯
╭─ SUMMARY: scale ───────────────────────────────────────────────────╮
│ Duration: 0.0131s │
│ Shape: 72 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● LAYERS │ ✚ scaled (array, 72x2000) │
│ │
╰────────────────────────────────────────────────────────────────────╯
computing PCA🔍
with n_comps=30
🖥️ Using sklearn PCA for CPU computation
🖥️ sklearn PCA backend: CPU computation
📊 PCA input data type: ArrayView, shape: (72, 2000), dtype: float64
🔧 PCA solver used: covariance_eigh
finished✅ (0.92s)
╭─ SUMMARY: pca ─────────────────────────────────────────────────────╮
│ Duration: 0.9311s │
│ Shape: 72 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ pca │
│ │ └─ params: {'zero_center': True, 'use_highly_variable': Tr...│
│ │ ✚ scaled|original|cum_sum_eigenvalues │
│ │ ✚ scaled|original|pca_var_ratios │
│ │
│ ● OBSM │ ✚ X_pca (array, 72x30) │
│ │ ✚ scaled|original|X_pca (array, 72x30) │
│ │
╰────────────────────────────────────────────────────────────────────╯
🖥️ Using Scanpy CPU to calculate neighbors...
🔍 K-Nearest Neighbors Graph Construction:
Mode: cpu
Neighbors: 15
Method: umap
Metric: euclidean
Representation: X_pca
🔍 Computing neighbor distances...
🔍 Computing connectivity matrix...
💡 Using UMAP-style connectivity
✓ Graph is fully connected
✅ KNN Graph Construction Completed Successfully!
✓ Processed: 72 cells with 15 neighbors each
✓ Results added to AnnData object:
• 'neighbors': Neighbors metadata (adata.uns)
• 'distances': Distance matrix (adata.obsp)
• 'connectivities': Connectivity matrix (adata.obsp)
╭─ SUMMARY: neighbors ───────────────────────────────────────────────╮
│ Duration: 0.138s │
│ Shape: 72 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ neighbors │
│ │ └─ params: {'n_neighbors': 15, 'method': 'umap', 'random_s...│
│ │
│ ● OBSP │ ✚ connectivities (sparse matrix, 72x72) │
│ │ ✚ distances (sparse matrix, 72x72) │
│ │
╰────────────────────────────────────────────────────────────────────╯
🔍 [2026-05-19 18:45:29] Running UMAP in 'cpu' mode...
🖥️ Using Scanpy CPU UMAP...
🔍 UMAP Dimensionality Reduction:
Mode: cpu
Method: umap
Components: 2
Min distance: 0.5
{'n_neighbors': 15, 'method': 'umap', 'random_state': 0, 'metric': 'euclidean', 'use_rep': 'X_pca'}
🔍 Computing UMAP parameters...
🔍 Computing UMAP embedding (classic method)...
✅ UMAP Dimensionality Reduction Completed Successfully!
✓ Embedding shape: 72 cells × 2 dimensions
✓ Results added to AnnData object:
• 'X_umap': UMAP coordinates (adata.obsm)
• 'umap': UMAP parameters (adata.uns)
✅ UMAP completed successfully.
╭─ SUMMARY: umap ────────────────────────────────────────────────────╮
│ Duration: 0.0097s │
│ Shape: 72 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ umap │
│ │ └─ params: {'a': 0.5830300199950147, 'b': 1.334166993228519}│
│ │
│ ● OBSM │ ✚ X_umap (array, 72x2) │
│ │
╰────────────────────────────────────────────────────────────────────╯
9. Downstream task 1 — differential expression#
Find marker genes per celltype on the metacell AnnData using
ov.single.find_markers (the omicverse Wilcoxon wrapper with pts=True
for per-cluster expression fractions).
# Drop celltypes with <2 metacells (find_markers needs n>=2 per group).
counts = ad_mc.obs['clusters'].value_counts()
keep = counts[counts >= 2].index.tolist()
ad_mc_de = ad_mc[ad_mc.obs['clusters'].isin(keep)].copy()
ad_mc_de.obs['clusters'] = ad_mc_de.obs['clusters'].astype('category')
ov.single.find_markers(ad_mc_de, groupby='clusters', method='wilcoxon',
key_added='rank_genes_groups', pts=True, use_gpu=False)
ov.single.get_markers(ad_mc_de, n_genes=3, key='rank_genes_groups')
🔍 Finding marker genes | method: wilcoxon | groupby: clusters | n_groups: 8 | n_genes: 50
✅ Done | 8 groups × 50 genes | corr: benjamini-hochberg | stored in adata.uns['rank_genes_groups']
| group | rank | names | scores | logfoldchanges | pvals | pvals_adj | pct_group | pct_rest | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | Alpha | 1 | Asb4 | 4.827149 | 5.818245 | 1.385013e-06 | 2.418402e-04 | 1.0 | 0.523810 |
| 1 | Alpha | 2 | Smarca1 | 4.776068 | 3.182478 | 1.787557e-06 | 2.418402e-04 | 1.0 | 1.000000 |
| 2 | Alpha | 3 | Ocrl | 4.776068 | 3.136611 | 1.787557e-06 | 2.418402e-04 | 1.0 | 0.984127 |
| 3 | Beta | 1 | Gng12 | 4.827149 | 3.927392 | 1.385013e-06 | 1.934722e-04 | 1.0 | 1.000000 |
| 4 | Beta | 2 | Sec61b | 4.827149 | 1.642527 | 1.385013e-06 | 1.934722e-04 | 1.0 | 1.000000 |
| 5 | Beta | 3 | Gm27033 | 4.827149 | 3.917109 | 1.385013e-06 | 1.934722e-04 | 1.0 | 0.952381 |
| 6 | Delta | 1 | Cd24a | 3.343364 | 2.544938 | 8.276928e-04 | 4.196597e-02 | 1.0 | 1.000000 |
| 7 | Delta | 2 | Spock3 | 3.343364 | 5.352329 | 8.276928e-04 | 4.196597e-02 | 1.0 | 0.750000 |
| 8 | Delta | 3 | Mest | 3.343364 | 4.213267 | 8.276928e-04 | 4.196597e-02 | 1.0 | 1.000000 |
| 9 | Ductal | 1 | Tkt | 5.927646 | 1.698946 | 3.073082e-09 | 1.241199e-07 | 1.0 | 1.000000 |
| 10 | Ductal | 2 | Proser2 | 5.927646 | 2.839478 | 3.073082e-09 | 1.241199e-07 | 1.0 | 0.982456 |
| 11 | Ductal | 3 | Nudt19 | 5.927646 | 3.226622 | 3.073082e-09 | 1.241199e-07 | 1.0 | 1.000000 |
| 12 | Epsilon | 1 | Txndc12 | 3.710407 | 1.429926 | 2.069260e-04 | 1.620964e-02 | 1.0 | 1.000000 |
| 13 | Epsilon | 2 | Foxd3 | 3.710407 | 8.726132 | 2.069260e-04 | 1.620964e-02 | 1.0 | 0.044776 |
| 14 | Epsilon | 3 | Gm11837 | 3.710407 | 5.789987 | 2.069260e-04 | 1.620964e-02 | 1.0 | 0.895522 |
| 15 | Ngn3 high EP | 1 | Cbfa2t3 | 6.054562 | 3.596581 | 1.408005e-09 | 2.708256e-07 | 1.0 | 0.946429 |
| 16 | Ngn3 high EP | 2 | Sh3bgrl3 | 6.054562 | 1.468390 | 1.408005e-09 | 2.708256e-07 | 1.0 | 1.000000 |
| 17 | Ngn3 high EP | 3 | Rnf114 | 6.041017 | 1.807264 | 1.531460e-09 | 2.708256e-07 | 1.0 | 1.000000 |
| 18 | Ngn3 low EP | 1 | Cited4 | 3.343364 | 2.892943 | 8.276928e-04 | 8.960757e-02 | 1.0 | 0.941176 |
| 19 | Ngn3 low EP | 2 | Cldn2 | 3.294197 | 4.624810 | 9.870337e-04 | 8.960757e-02 | 1.0 | 0.588235 |
| 20 | Ngn3 low EP | 3 | Ascl1 | 3.269613 | 4.734896 | 1.076946e-03 | 8.960757e-02 | 1.0 | 0.588235 |
| 21 | Pre-endocrine | 1 | Eif3e | 5.015152 | 0.868462 | 5.299167e-07 | 1.744158e-04 | 1.0 | 1.000000 |
| 22 | Pre-endocrine | 2 | Cystm1 | 4.966303 | 1.284075 | 6.824141e-07 | 1.744158e-04 | 1.0 | 1.000000 |
| 23 | Pre-endocrine | 3 | Foxp1 | 4.950020 | 1.440488 | 7.420595e-07 | 1.744158e-04 | 1.0 | 1.000000 |
10. Downstream task 2 — marker dotplot#
ov.pl.markers_dotplot reads the rank_genes_groups result and shows the
top-N markers per group with both expression intensity (colour) and the
fraction of metacells in which each gene is expressed (dot size).
Canonical pancreas markers (Ins1/Ins2 for Beta, Gcg for Alpha, etc.)
should pop out clearly even on this small metacell set.
ov.pl.markers_dotplot(ad_mc_de, groupby='clusters', n_genes=3,
key='rank_genes_groups')

11. Save the metacell partition#
Save the slim state (assignments + soft membership + config). The
companion load recovers the unified AnnData schema and lets you re-run
predicted() / compute_purity() / etc. without re-fitting.
import tempfile, os
with tempfile.NamedTemporaryFile(suffix='.pkl', delete=False) as f:
path = f.name
mc.save(path)
print(f'saved to {path}')
os.remove(path)
saved to /tmp/tmpet9z7zry.pkl
12. Next steps#
Multi-sample data? Move on to t_metacell_multisample — same workflow but with batch correction first so per-sample metacells live in a shared embedding.
Need out-of-sample assignment (new cells arrive over time)? Switch the backend to
metaqand usemc.assign_new_cells(adata_new)— see zoo/t_metacell_metaq.Want to validate the choice of backend? Run
ov.single.compare_metacell_backendson your data — see zoo/t_metacell_compare.Want to use metacells in cell–cell communication / SCENIC? Pass
ad_mc(the AnnData returned bymc.predicted()) into the standardov.single.pCellPhoneDB,ov.single.pySCENIC, etc. workflows — they consume the unified schema directly.