Differential correlation — DGCA#
Classical differential analysis (metabol.differential) asks which
metabolites change in abundance? Differential correlation asks a
complementary question: which metabolite–metabolite relationships
change between conditions? Two metabolites might have identical mean
levels in both groups yet be tightly co-regulated in one and
uncorrelated in the other — evidence for a rewired pathway.
ov.metabol.dgca implements DGCA (McKenzie et al., BMC
Genomics 2016): Fisher-z transformation of each pair’s correlation
in each condition, z-test on the difference, BH FDR, and a
categorical DC class label (+/+, +/0, +/-, …).
We use the Cachexia dataset — 77 urinary metabolomes labelled
cachexic vs control (47 vs 30).
0 — Setup#
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import omicverse as ov
csv_path = ov.datasets.download_data(
url='https://rest.xialab.ca/api/download/metaboanalyst/human_cachexia.csv',
file_path='human_cachexia.csv',
dir='metabol_demo',
)
adata = ov.metabol.read_metaboanalyst(csv_path, group_col='Muscle loss')
adata = ov.metabol.impute(adata, method='qrilc', seed=0)
adata = ov.metabol.normalize(adata, method='pqn')
adata = ov.metabol.transform(adata, method='log')
adata
🔍 Downloading data to metabol_demo/human_cachexia.csv
⚠️ File metabol_demo/human_cachexia.csv already exists
AnnData object with n_obs × n_vars = 77 × 63
obs: 'group'
var: 'missing_frac'
uns: 'metabol'
layers: 'raw'
1 — Run DGCA#
For 63 metabolites we get 63*62/2 = 1953 pairs. Use Spearman for
metabolomics (heavy-tailed distributions) and threshold |r|≥0.3 for
class labels (the default, matching MetaboAnalyst’s network module).
dc = ov.metabol.dgca(
adata,
group_col='group',
group_a='cachexic', group_b='control',
method='spearman',
abs_r_threshold=0.3,
)
print(f'{len(dc)} pairs tested')
dc.head(15)
1953 pairs tested
feature_a feature_b r_a r_b z_diff \
0 Citrate Glycine 0.351989 0.856285 -3.728665
1 Dimethylamine Succinate 0.450278 -0.410456 3.768224
2 Lactate Taurine 0.078978 -0.713014 3.977756
3 2-Oxoglutarate Glutamine -0.019832 0.666741 -3.373402
4 3-Indoxylsulfate Guanidoacetate 0.296369 -0.472303 3.348357
5 Histidine Trigonelline 0.209644 -0.531924 3.295424
6 Guanidoacetate tau-Methylhistidine 0.346438 -0.407341 3.247102
7 Acetone Tyrosine 0.286193 -0.421580 3.043459
8 Betaine Ethanolamine -0.257285 0.450945 -3.064152
9 Methylamine Methylguanidine 0.272036 -0.434483 3.045276
10 Ethanolamine cis-Aconitate 0.463344 -0.228921 3.004948
11 Alanine Tyrosine 0.065796 0.653838 -2.929152
12 Asparagine N,N-Dimethylglycine -0.254170 0.426029 -2.924320
13 Glucose Histidine 0.074931 -0.568854 2.948854
14 1-Methylnicotinamide Trigonelline 0.011506 -0.580868 2.762240
pvalue padj dc_class
0 0.000192 0.125315 +/+
1 0.000164 0.125315 +/-
2 0.000070 0.125315 0/-
3 0.000742 0.317527 0/+
4 0.000813 0.317527 0/-
5 0.000983 0.319879 0/-
6 0.001166 0.325277 +/-
7 0.002339 0.456759 0/-
8 0.002183 0.456759 0/+
9 0.002325 0.456759 0/-
10 0.002656 0.471608 +/0
11 0.003399 0.481568 0/+
12 0.003452 0.481568 0/+
13 0.003190 0.481568 0/-
14 0.005741 0.509612 0/-
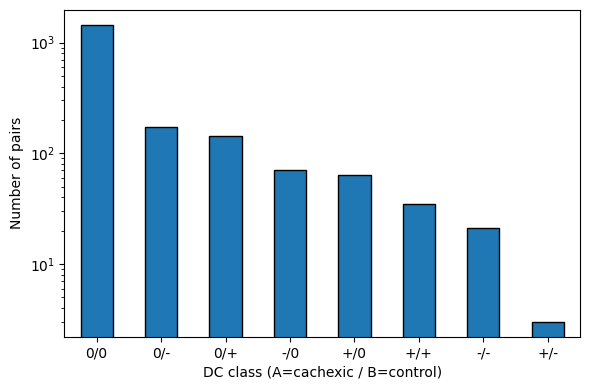
2 — DC class distribution#
Each pair gets a two-symbol label A/B where +/-/0 summarise the
correlation strength in groups cachexic (A) and control (B):
class_counts = dc['dc_class'].value_counts()
class_counts
dc_class
0/0 1444
0/- 173
0/+ 143
-/0 71
+/0 63
+/+ 35
-/- 21
+/- 3
Name: count, dtype: int64
fig, ax = plt.subplots(figsize=(6, 4))
class_counts.plot.bar(ax=ax, color='C0', edgecolor='k')
ax.set_ylabel('Number of pairs')
ax.set_xlabel('DC class (A=cachexic / B=control)')
ax.set_yscale('log')
plt.xticks(rotation=0)
fig.tight_layout()
plt.show()
3 — Top rewired pairs#
Sort by |z_diff| and inspect the most differentially correlated pairs.
Cachexia (77 samples) has limited power for pairwise inference — with
BH FDR at 5% only strong effects survive; we therefore rank by effect
size rather than filter on padj so the notebook illustrates the
API regardless.
dc_top = dc.copy()
dc_top['abs_z'] = dc_top['z_diff'].abs()
top = dc_top.sort_values('abs_z', ascending=False).head(15)
top[['feature_a', 'feature_b', 'r_a', 'r_b', 'z_diff', 'padj', 'dc_class']]
feature_a feature_b r_a r_b z_diff \
2 Lactate Taurine 0.078978 -0.713014 3.977756
1 Dimethylamine Succinate 0.450278 -0.410456 3.768224
0 Citrate Glycine 0.351989 0.856285 -3.728665
3 2-Oxoglutarate Glutamine -0.019832 0.666741 -3.373402
4 3-Indoxylsulfate Guanidoacetate 0.296369 -0.472303 3.348357
5 Histidine Trigonelline 0.209644 -0.531924 3.295424
6 Guanidoacetate tau-Methylhistidine 0.346438 -0.407341 3.247102
8 Betaine Ethanolamine -0.257285 0.450945 -3.064152
9 Methylamine Methylguanidine 0.272036 -0.434483 3.045276
7 Acetone Tyrosine 0.286193 -0.421580 3.043459
10 Ethanolamine cis-Aconitate 0.463344 -0.228921 3.004948
13 Glucose Histidine 0.074931 -0.568854 2.948854
11 Alanine Tyrosine 0.065796 0.653838 -2.929152
12 Asparagine N,N-Dimethylglycine -0.254170 0.426029 -2.924320
21 Pyroglutamate Succinate 0.503469 -0.149722 2.882995
padj dc_class
2 0.125315 0/-
1 0.125315 +/-
0 0.125315 +/+
3 0.317527 0/+
4 0.317527 0/-
5 0.319879 0/-
6 0.325277 +/-
8 0.456759 0/+
9 0.456759 0/-
7 0.456759 0/-
10 0.471608 +/0
13 0.481568 0/-
11 0.481568 0/+
12 0.481568 0/+
21 0.509612 +/0
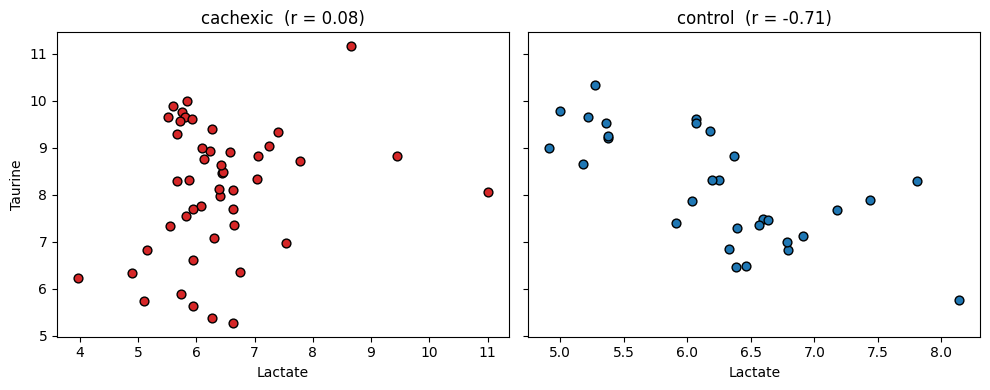
4 — Visualise one rewired pair#
Pick the top pair and show the raw correlation in each group on a scatter plot:
top_row = top.iloc[0]
fa, fb = top_row['feature_a'], top_row['feature_b']
idx_a = list(adata.var_names).index(fa)
idx_b = list(adata.var_names).index(fb)
groups = adata.obs['group'].astype(str).to_numpy()
X = np.asarray(adata.X)
fig, axes = plt.subplots(1, 2, figsize=(10, 4), sharey=True)
for ax, label, colour in zip(axes, ['cachexic', 'control'], ['C3', 'C0']):
mask = groups == label
ax.scatter(X[mask, idx_a], X[mask, idx_b], color=colour,
edgecolor='k', s=40)
ax.set_xlabel(fa)
ax.set_title(f'{label} (r = {(top_row["r_a"] if label == "cachexic" else top_row["r_b"]):.2f})')
axes[0].set_ylabel(fb)
fig.tight_layout()
plt.show()
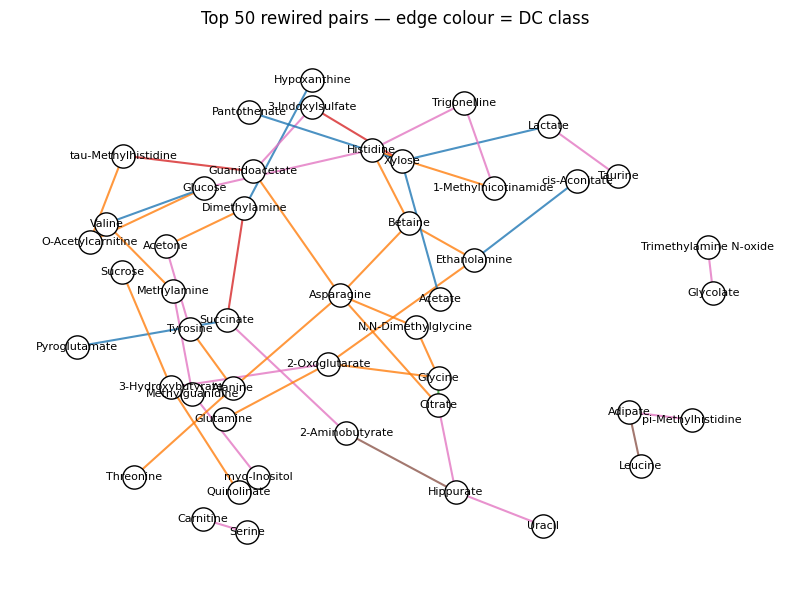
5 — Rewired correlation network#
Build a small NetworkX graph of the top-rewired pairs coloured by DC class:
import networkx as nx
top50 = dc_top.sort_values('abs_z', ascending=False).head(50)
G = nx.Graph()
class_colour = {'+/+': 'C2', '-/-': 'C4', '+/-': 'C3', '-/+': 'C3',
'+/0': 'C0', '0/+': 'C1', '-/0': 'C5', '0/-': 'C6', '0/0': 'lightgray'}
for _, r in top50.iterrows():
G.add_edge(r['feature_a'], r['feature_b'],
weight=abs(r['z_diff']),
color=class_colour.get(r['dc_class'], 'gray'))
pos = nx.spring_layout(G, seed=0, k=0.7)
edge_colors = [G[u][v]['color'] for u, v in G.edges()]
fig, ax = plt.subplots(figsize=(8, 6))
nx.draw_networkx_edges(G, pos, edge_color=edge_colors, width=1.5, alpha=0.8)
nx.draw_networkx_nodes(G, pos, node_size=280, node_color='white',
edgecolors='k', ax=ax)
nx.draw_networkx_labels(G, pos, font_size=8, ax=ax)
ax.set_title('Top 50 rewired pairs — edge colour = DC class')
ax.axis('off')
fig.tight_layout()
plt.show()
6 — Static per-condition networks (corr_network)#
DGCA compares conditions. Sometimes you want the static network in each condition on its own — a co-regulation backbone you can visualise, annotate with pathways, or export to Cytoscape.
ov.metabol.corr_network returns a filtered edge list (|r| ≥
threshold, padj < α) for a single condition. Below we build networks
separately for cachexic and control and compare their size / overlap:
edges_ca = ov.metabol.corr_network(
adata,
group_col='group', group='cachexic',
method='spearman',
abs_r_threshold=0.5, padj_threshold=0.05,
)
edges_ct = ov.metabol.corr_network(
adata,
group_col='group', group='control',
method='spearman',
abs_r_threshold=0.5, padj_threshold=0.05,
)
print(f'cachexic network : {len(edges_ca)} edges on {edges_ca.attrs["n_samples"]} samples')
print(f'control network : {len(edges_ct)} edges on {edges_ct.attrs["n_samples"]} samples')
def pair_key(row):
return frozenset((row['feature_a'], row['feature_b']))
ca_pairs = set(edges_ca.apply(pair_key, axis=1))
ct_pairs = set(edges_ct.apply(pair_key, axis=1))
print(f'shared edges : {len(ca_pairs & ct_pairs)}')
print(f'cachexic-only : {len(ca_pairs - ct_pairs)}')
print(f'control-only : {len(ct_pairs - ca_pairs)}')
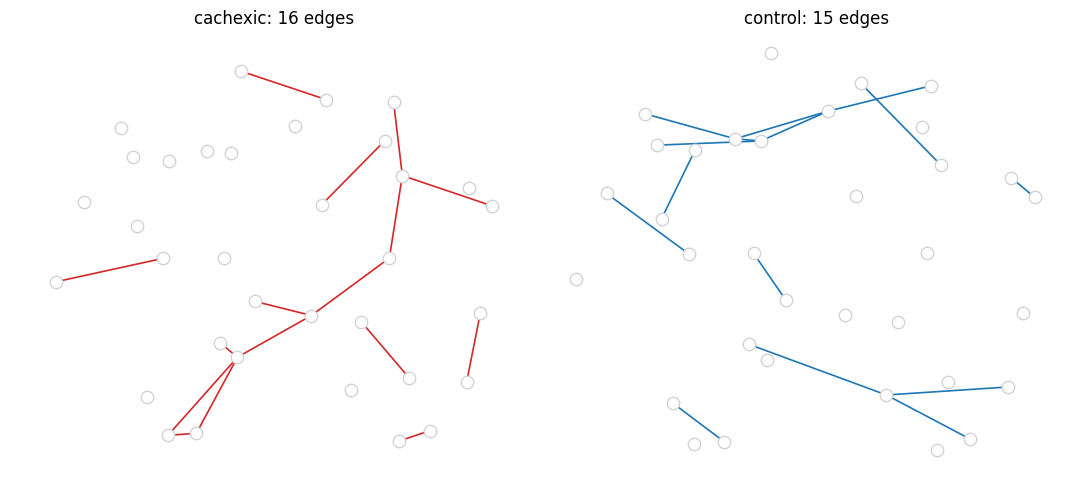
cachexic network : 16 edges on 47 samples
control network : 15 edges on 30 samples
shared edges : 0
cachexic-only : 16
control-only : 15
Top edges in the cachexic network#
edges_ca.head(10)
feature_a feature_b r pvalue \
0 Creatinine Dimethylamine 0.768270 1.583932e-11
1 Carnitine O-Acetylcarnitine 0.662812 1.209255e-07
2 Glucose Lactate 0.609389 2.652975e-06
3 Acetate Succinate 0.606614 3.059246e-06
4 Dimethylamine Trimethylamine N-oxide 0.569380 1.794035e-05
5 Dimethylamine Pyroglutamate 0.564870 2.185967e-05
6 2-Oxoglutarate Acetate -0.561748 2.501540e-05
7 Creatinine Hypoxanthine 0.550994 3.933656e-05
8 pi-Methylhistidine tau-Methylhistidine 0.526364 1.039705e-04
9 Acetate Methylguanidine -0.520467 1.295807e-04
padj
0 3.093420e-08
1 1.180838e-04
2 1.493677e-03
3 1.493677e-03
4 6.979296e-03
5 6.979296e-03
6 6.979296e-03
7 9.603037e-03
8 2.256160e-02
9 2.530712e-02
Venn-like side-by-side#
Plot the two networks with a common layout (union of nodes) so differences in who connects to whom become obvious.
import networkx as nx
all_nodes = list(set(edges_ca['feature_a']) | set(edges_ca['feature_b'])
| set(edges_ct['feature_a']) | set(edges_ct['feature_b']))
Gu = nx.Graph()
Gu.add_nodes_from(all_nodes)
for df in (edges_ca, edges_ct):
for _, r in df.iterrows():
Gu.add_edge(r['feature_a'], r['feature_b'])
pos = nx.spring_layout(Gu, seed=0, k=0.6)
fig, axes = plt.subplots(1, 2, figsize=(11, 5))
for ax, edges, title, colour in zip(
axes, [edges_ca, edges_ct],
['cachexic', 'control'],
['C3', 'C0']):
G = nx.from_pandas_edgelist(edges,
source='feature_a', target='feature_b',
edge_attr='r')
nx.draw_networkx_nodes(Gu, pos, node_size=80,
node_color='white', edgecolors='lightgray', ax=ax)
nx.draw_networkx_edges(G, pos, edge_color=colour, width=1.2, ax=ax)
ax.set_title(f'{title}: {G.number_of_edges()} edges')
ax.axis('off')
fig.tight_layout()
plt.show()
Takeaways#
DGCA surfaces relationships that
metabol.differentialmisses.The class labels (
+/+,+/0,+/-…) give an interpretable summary:+/0= “correlation gained in A”,+/-= “inversion”.For large panels (
p > 500) restrict withfeatures=<top-N by univariate AUC or VIP>to keep the O(p²) output manageable.
DGCA pairs naturally with ASCA + biomarker panels: they answer different questions about the same data.