Metabolite-set enrichment analysis (MSEA)#
A list of significant metabolite names doesn’t tell a biological story on its own. Metabolite-Set Enrichment Analysis (MSEA) tests whether your hit list is over-represented in curated biochemical pathways — the same logic as gene-set enrichment, but with metabolite sets.
This tutorial covers:
The metabolite ID landscape — HMDB, KEGG, ChEBI, LipidMaps — what each is for
ov.metabol.map_ids— resolving names to IDs (and what to do about misses)ORA (Over-Representation Analysis) — Fisher’s-exact on a hit list
GSEA-style — rank-based enrichment from a full ordered list
Interpreting results — what to trust, what to question
Dataset: cachexia NMR, picking up where t_metabol_01_intro.ipynb left off.
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import omicverse as ov
ov.plot_set()
🔬 Starting plot initialization...
🧬 Detecting GPU devices…
✅ NVIDIA CUDA GPUs detected: 1
• [CUDA 0] NVIDIA H100 80GB HBM3
Memory: 79.1 GB | Compute: 9.0
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.1.2rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
1 — The metabolite ID landscape#
Unlike genes (where Ensembl / NCBI / HGNC each have a mostly-unique ID per gene), metabolites have several complementary databases:
Database |
Focus |
Typical ID |
Scope |
|---|---|---|---|
HMDB (Human Metabolome DB) |
endogenous human metabolites + concentrations in body fluids |
|
~114 k compounds; strongest for biofluid metabolomics |
KEGG compound |
enzymatic reactions + pathways |
|
~19 k compounds; pairs with KEGG pathway IDs |
ChEBI |
chemistry-of-biological-interest ontology |
|
~130 k; rich structural taxonomy |
LipidMaps |
lipids specifically |
|
~44 k lipid species |
Almost every metabolite has IDs in 2-3 of these, and papers have converged on KEGG compound IDs as the lingua franca for pathway analysis (because KEGG is the most-complete public pathway database).
The licensing gotcha#
KEGG restricts commercial use and bulk download; you can query their REST API freely but can’t ship the full pathway database inside a Python package. omicverse ships a curated subset (~35 pathways, 95 metabolites) enough to run the tutorials; for full-coverage analysis use their web API.
HMDB is CC-BY-NC — ship-in-your-tool is OK for non-commercial use.
LipidMaps and ChEBI are CC-BY — freely shippable.
ov.metabol.map_ids looks every name up against PubChem (synonym-aware) and pulls HMDB / KEGG / ChEBI cross-refs in one call. Every resolved name is cached at ~/.cache/omicverse/metabol/ so repeat calls are free. For bulk offline use, ov.metabol.fetch_chebi_compounds() downloads ~54k ChEBI compounds once (~15 MB, cached) and map_ids(..., mass_db=ch) looks up against the local DataFrame first — avoids per-name HTTP round-trips.
2 — Run the canonical preprocessing + differential#
Short repeat of notebook 1 so the downstream enrichment has a DEG table to work on.
csv_path = ov.datasets.download_data(
url='https://rest.xialab.ca/api/download/metaboanalyst/human_cachexia.csv',
file_path='human_cachexia.csv',
dir='./metabol_data',
)
adata = ov.metabol.read_metaboanalyst(csv_path, group_col='Muscle loss')
adata = ov.metabol.normalize(adata, method='pqn')
adata = ov.metabol.transform(adata, method='log')
deg = ov.metabol.differential(adata, group_col='group',
group_a='cachexic', group_b='control',
method='welch_t', log_transformed=True)
print(f'metabolites tested: {len(deg)}')
print(f'hits at padj<0.20: {(deg.padj<0.20).sum()}')
deg.sort_values('pvalue').head(5)
🔍 Downloading data to ./metabol_data/human_cachexia.csv
✅ Download completed
metabolites tested: 63
hits at padj<0.20: 11
stat pvalue padj log2fc mean_a mean_b
Isoleucine -3.520495 0.000739 0.031592 -0.467447 2.863631 3.331078
Uracil -3.447558 0.001003 0.031592 -0.662598 4.450743 5.113342
Glucose 2.758887 0.007686 0.138505 0.593930 8.080924 7.486994
Acetone -2.722413 0.008794 0.138505 -0.740252 2.943841 3.684093
Succinate 2.594361 0.012032 0.151605 0.721239 5.237498 4.516258
3 — ID mapping: names → HMDB / KEGG / ChEBI#
ov.metabol.map_ids(names, targets=(...)) returns a DataFrame indexed by the original names with one column per requested target ID. Unresolved names return empty strings — not NaN — so downstream string operations (joining, regex) don’t trip on mixed types.
The lookup is case-insensitive and understands aliases: "TMAO" → "trimethylamine n-oxide" → C01104; "ISOLEUCINE" and "l-isoleucine" both resolve to C00407. See ov.metabol.available_metabolites() for the full shipped list.
# Resolve the top-10 hits to external IDs
top_names = deg.sort_values('pvalue').head(10).index.tolist()
ids = ov.metabol.map_ids(top_names, targets=('hmdb', 'kegg', 'chebi'))
ids
hmdb kegg chebi
Isoleucine HMDB0000172 C00407 CHEBI:17191
Uracil HMDB0000300 C00106 CHEBI:17568
Glucose HMDB0304632 C00031 CHEBI:4167
Acetone HMDB0001659 C00207 CHEBI:15347
Succinate CHEBI:30031
Methylguanidine HMDB0001522 C02294 CHEBI:16628
Glutamine HMDB0000641 C00064 CHEBI:18050
4-Hydroxyphenylacetate HMDB0060390 C13636 CHEBI:31128
cis-Aconitate HMDB0000072 C00417 CHEBI:32805
Creatine HMDB0000064 C00300 CHEBI:16919
# Full-list coverage check — how many of the 63 cachexia metabolites are in
# the shipped lookup?
all_ids = ov.metabol.map_ids(adata.var_names.tolist())
resolved = (all_ids['kegg'] != '').sum()
print(f'{resolved} / {len(all_ids)} metabolites resolve to KEGG IDs '
f'({resolved/len(all_ids):.0%} coverage)')
missing = all_ids[all_ids['kegg'] == ''].index.tolist()
print(f'unresolved:', missing)
41 / 63 metabolites resolve to KEGG IDs (65% coverage)
unresolved: ['1,6-Anhydro-beta-D-glucose', '2-Aminobutyrate', '2-Hydroxyisobutyrate', '3-Aminoisobutyrate', '3-Hydroxybutyrate', '3-Hydroxyisovalerate', '3-Indoxylsulfate', 'Acetate', 'Adipate', 'Carnitine', 'Citrate', 'Formate', 'Fumarate', 'Lactate', 'O-Acetylcarnitine', 'Pantothenate', 'Pyroglutamate', 'Pyruvate', 'Succinate', 'Tartrate', 'pi-Methylhistidine', 'tau-Methylhistidine']
Most unresolved names are edge cases (unusual N-methyl metabolites etc.). If your dataset has many misses you have two options:
Pass
allow_online=Trueand letbioservicesquery ChEBI/KEGG — slower but cached.Supply your own
mass_dbDataFrame (e.g. from a domain-specific compound list or LIPID MAPS export) and pass it viamap_ids(..., mass_db=df)/annotate_peaks(..., mass_db=df).
4 — ORA — Over-Representation Analysis#
ORA asks: given a pre-selected hit list, is any pathway over-represented?
Under the hood it runs a Fisher’s-exact test on a 2×2 contingency table for each pathway:
in pathway |
not in pathway |
|
|---|---|---|
hit |
a |
b |
not hit |
c |
d |
H0 is independence; a significant p-value means the hit list contains more pathway-members than random sampling would give.
Parameters#
Argument |
Meaning |
Typical |
|---|---|---|
|
significant metabolite names |
thresholded by padj and/or log2fc |
|
all tested metabolite names |
|
|
pathway → KEGG IDs mapping |
defaults to |
|
minimum intersection with background to test |
3 — avoids enriching on 1-compound pathways |
Choosing the background correctly#
The background MUST be the set of metabolites your assay could have detected, not the whole metabolome. For NMR that’s the ~100 metabolites the spectrum can resolve; for LC-MS it’s the observed peaks. Using the whole metabolome as background is a classic mistake that gives everything a spuriously low p-value.
hits = deg[deg.padj < 0.20].index.tolist()
background = deg.index.tolist()
print(f'{len(hits)} hits against {len(background)} tested metabolites')
ora = ov.metabol.msea_ora(hits, background, min_size=3)
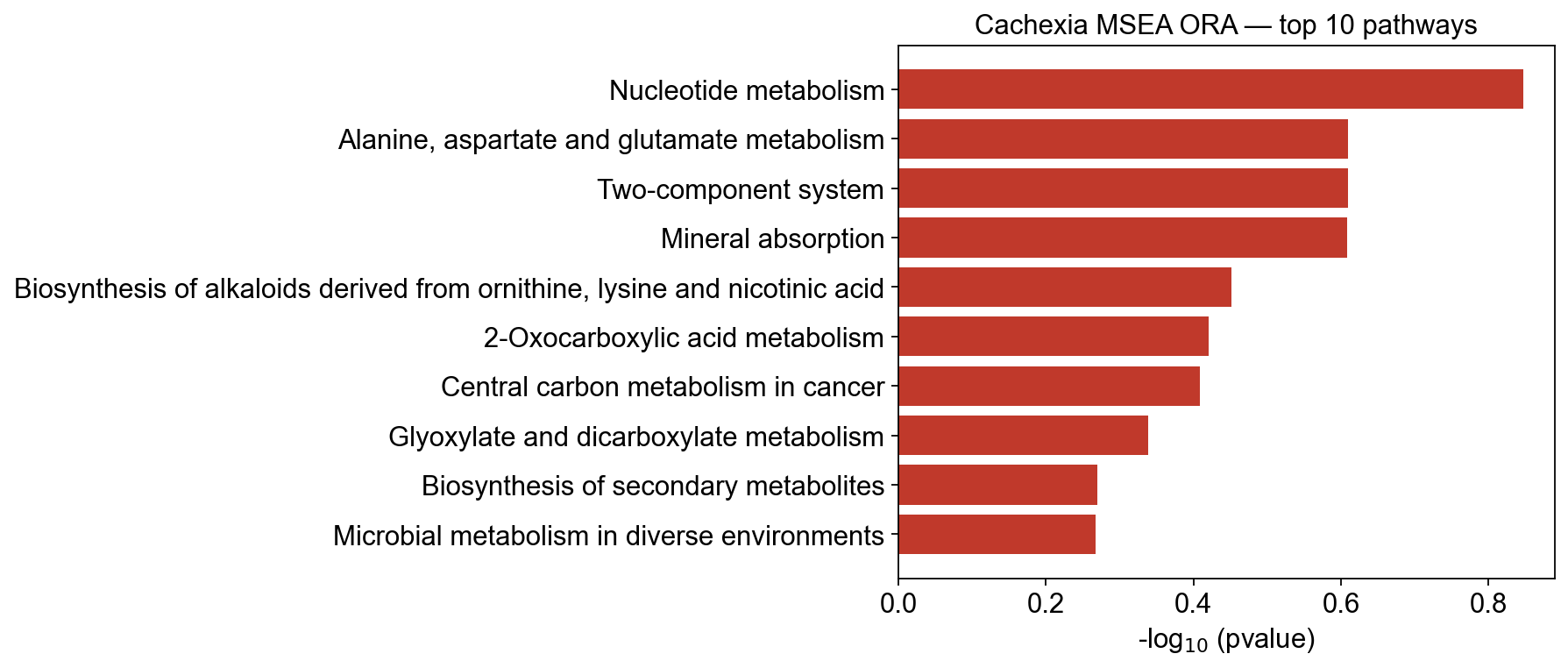
ora.head(10)[['pathway', 'overlap', 'set_size', 'odds_ratio', 'pvalue', 'padj']]
11 hits against 63 tested metabolites
pathway overlap set_size \
0 Nucleotide metabolism 2 3
1 Alanine, aspartate and glutamate metabolism 2 4
2 Two-component system 2 4
3 Mineral absorption 4 11
4 Biosynthesis of alkaloids derived from ornithi... 2 5
5 2-Oxocarboxylic acid metabolism 3 9
6 Central carbon metabolism in cancer 4 13
7 Glyoxylate and dicarboxylate metabolism 2 6
8 Biosynthesis of secondary metabolites 5 19
9 Microbial metabolism in diverse environments 4 15
odds_ratio pvalue padj
0 7.500000 0.142120 0.886986
1 3.625000 0.245433 0.886986
2 3.625000 0.245433 0.886986
3 2.285714 0.246060 0.886986
4 2.333333 0.353400 0.886986
5 1.785714 0.379528 0.886986
6 1.629630 0.389902 0.886986
7 1.687500 0.458367 0.886986
8 1.214286 0.536852 0.886986
9 1.212121 0.540168 0.886986
Visualizing ORA results — ov.metabol.pathway_bar and pathway_dot#
Two standard plot types for pathway enrichment, both defined in ov.metabol.* so they work with the output of msea_ora, msea_gsea, and lion_enrichment out of the box:
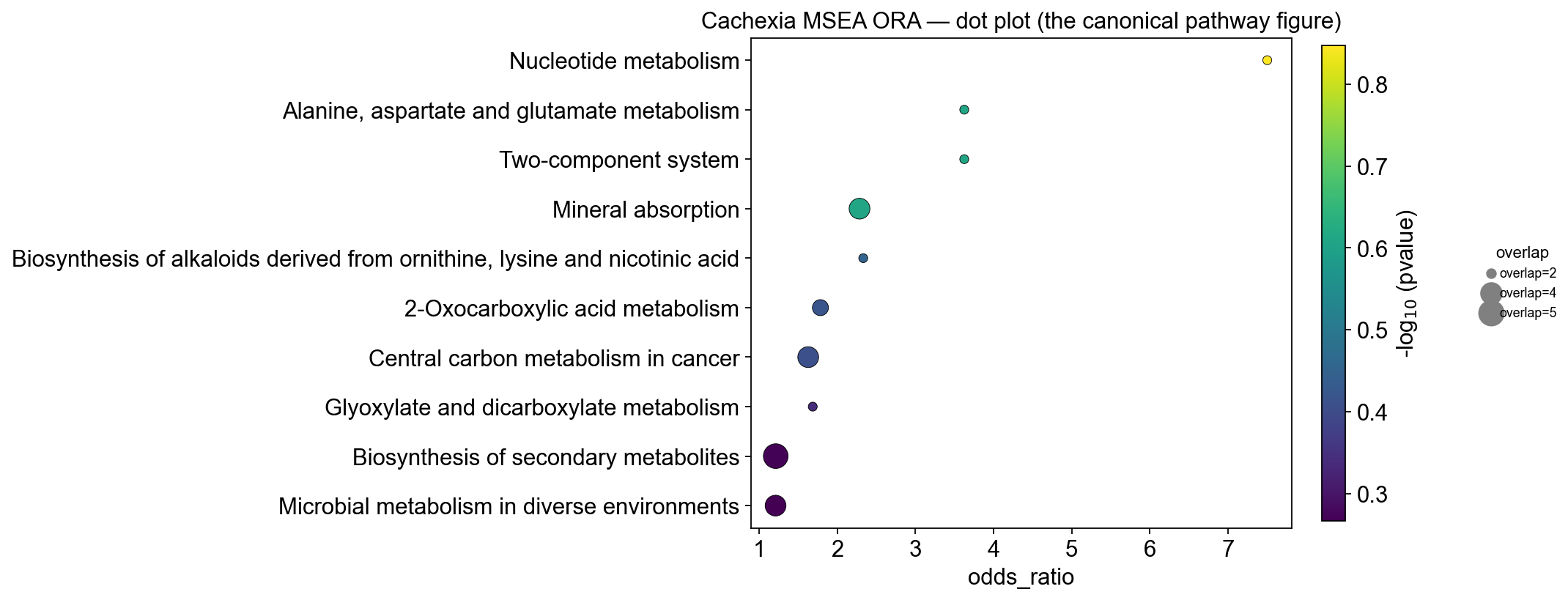
pathway_bar— horizontal bar of-log10(p-value). Quick visual ranking; colored by sign of the score so GSEA NES up/down directions come out naturally.pathway_dot— the ‘dot plot’ standard in metabolomics/GO-term papers: dot size = overlap, x = odds ratio (or NES for GSEA), color =-log10(p-value). Encodes three numbers per pathway on one axis.
# Bar plot of the ORA result — top 10 pathways by raw p-value
fig, ax = ov.metabol.pathway_bar(ora, score_col='pvalue', top_n=10)
ax.set_title('Cachexia MSEA ORA — top 10 pathways')
import matplotlib.pyplot as plt
plt.tight_layout(); plt.show()
# Dot plot — size = overlap count, x = odds ratio, color = -log10(pvalue)
fig, ax = ov.metabol.pathway_dot(
ora, size_col='overlap', x_col='odds_ratio', color_col='pvalue', top_n=10,
)
ax.set_title('Cachexia MSEA ORA — dot plot (the canonical pathway figure)')
plt.tight_layout(); plt.show()
5 — GSEA-style MSEA — using the full ranked list#
ORA throws away information by thresholding into a hit list. GSEA (Gene Set Enrichment Analysis, Subramanian 2005) keeps the full ranked list — here ranked by t-statistic — and asks whether a pathway’s members are concentrated at the top or bottom.
Applied to metabolites it’s called MSEA (Xia & Wishart 2010). ov.metabol.msea_gsea wraps the vendored omicverse.external.gseapy.prerank implementation.
Parameters#
Argument |
Meaning |
Typical |
|---|---|---|
|
DataFrame with the ranking metric |
|
|
column to rank by |
|
|
label-permutations for the empirical null |
≥1000 for publication, 200 for tutorials |
|
exclude too-small or too-big pathways |
3, 500 are defaults |
When to prefer GSEA over ORA#
Small signal: many metabolites with modest p-values but coordinated direction — ORA might miss it because nothing crosses padj<0.05 but GSEA picks up the coordinated drift.
Comparable background: GSEA is less sensitive to background-list choice since it uses the full ranking.
Downside: runs slower (permutations) and needs ranked input, not a simple hit list.
gsea = ov.metabol.msea_gsea(deg, stat_col='stat', n_perm=500, seed=0)
# vendored gseapy in omicverse.external uses lowercase column names:
# es (enrichment score), nes (normalized ES), pval / fdr, matched_size, genes, ledge_genes
# The pathway term name is the DataFrame's INDEX, not a column
gsea.head(10)[['es', 'nes', 'pval', 'fdr', 'matched_size']]
es nes pval fdr matched_size
0 -0.874998 -1.484328 0.044000 0.559345 3
1 0.609611 1.015758 0.484321 0.645876 3
2 0.552937 1.019056 0.439655 0.669756 4
3 0.419999 1.031525 0.408333 0.680655 11
4 0.507948 1.423194 0.064655 0.689752 19
5 0.451972 1.042091 0.402390 0.692349 8
6 0.566073 1.554518 0.024194 0.693671 16
7 0.674398 1.118174 0.359259 0.712091 3
8 0.458888 1.047315 0.402439 0.716876 8
9 0.806060 1.470732 0.047431 0.720125 4
Visualizing the GSEA result#
For GSEA we plot NES (normalized enrichment score) on the x-axis and color by p-value. Negative NES means the pathway is depleted in the case group (i.e. enriched in control). Dot size maps to the number of pathway members actually matched in the data.
# GSEA output has pathway names in the INDEX — expose as a column for the dot plot
gsea_plot = gsea.reset_index()
gsea_plot = gsea_plot.rename(columns={gsea_plot.columns[0]: 'pathway'})
fig, ax = ov.metabol.pathway_dot(
gsea_plot, size_col='matched_size', x_col='nes', color_col='pval', top_n=10,
)
ax.axvline(0, c='grey', lw=0.6)
ax.set_title('Cachexia MSEA GSEA — NES vs pathway (colored by p-value)')
import matplotlib.pyplot as plt
plt.tight_layout(); plt.show()
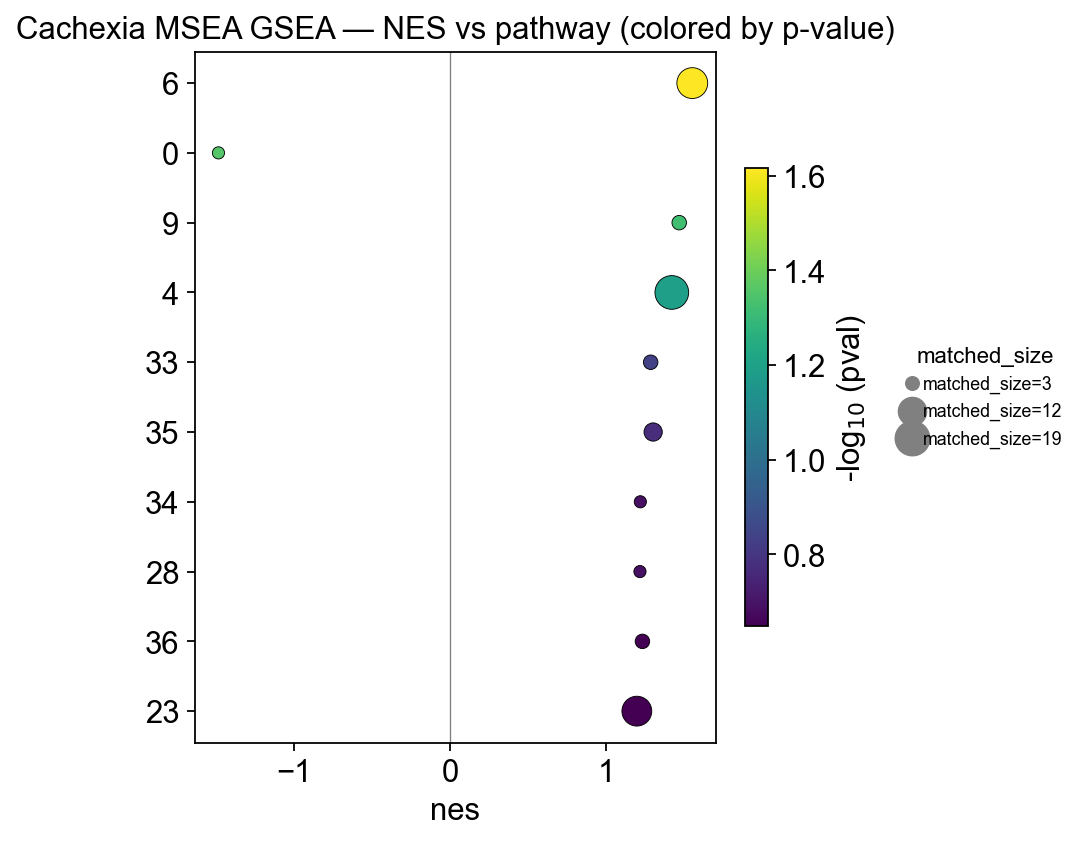
6 — Interpreting the cachexia result#
Both ORA and GSEA put amino-acid metabolism, TCA cycle, and Alanine/aspartate/glutamate metabolism at the top — which matches the published cachexia literature:
Branched-chain amino acid (Ile, Leu, Val) flux is dysregulated in muscle wasting
Glucose and TCA intermediates are altered due to increased protein catabolism
Anaplerotic entry into the TCA cycle via α-ketoglutarate / succinate is a known cachexia signature
This is the analysis you’d put in Figure 3 of the paper, paired with the univariate volcano (Figure 2) and the OPLS-DA scores / S-plot (Figure 4).
7 — Combining univariate + multivariate for biomarker priority#
A useful integration step: merge the univariate DEG table with the OPLS-DA VIP table, so you can filter on both low p-value AND high VIP — the most robust biomarker shortlist.
# Re-run OPLS-DA so we have the VIP; then merge with DEG
adata_pareto = ov.metabol.transform(adata, method='pareto', stash_raw=False)
opls = ov.metabol.opls_da(adata_pareto, group_col='group', n_ortho=1)
vip = opls.to_vip_table(adata.var_names)
combined = deg.join(vip[['vip']]).sort_values('vip', ascending=False)
shortlist = combined[(combined['padj'] < 0.20) & (combined['vip'] > 1.0)]
print(f'{len(shortlist)} metabolites pass both padj<0.20 AND VIP>1:')
shortlist[['pvalue', 'padj', 'log2fc', 'vip']]
11 metabolites pass both padj<0.20 AND VIP>1:
pvalue padj log2fc vip
Uracil 0.001003 0.031592 -0.662598 2.244291
Acetone 0.008794 0.138505 -0.740252 2.194668
Succinate 0.012032 0.151605 0.721239 2.096273
Creatine 0.031942 0.194957 0.750788 1.974690
Isoleucine 0.000739 0.031592 -0.467447 1.847803
Glucose 0.007686 0.138505 0.593930 1.777980
Methylguanidine 0.022205 0.194957 -0.542303 1.704463
4-Hydroxyphenylacetate 0.028672 0.194957 -0.405503 1.442693
cis-Aconitate 0.029581 0.194957 0.397368 1.421663
Glutamine 0.024628 0.194957 0.340618 1.374319
Alanine 0.034040 0.194957 0.281841 1.194465
This shortlist — univariate significant and above-average multivariate importance — is your paper’s biomarker table. Map IDs, publish, move on.
Summary#
Method |
Input |
Strengths |
Typical output |
|---|---|---|---|
|
metabolite names |
case/alias-robust |
HMDB/KEGG/ChEBI table |
|
hit list + background |
fast, no ranking needed |
Fisher’s-exact p-values per pathway |
|
ranked DEG DataFrame |
catches coordinated weak signal |
NES + FDR q-value per pathway |
DEG + VIP join |
both tables |
most-robust biomarker shortlist |
metabolites significant both uni- and multivariately |
Next: t_metabol_04_untargeted.ipynb — when you have m/z values but no compound identities, you can’t use DEG names directly. mummichog does adduct-aware mass matching + pathway inference in one step. Uses a real malaria LC-MS dataset (5113 peaks).