Consensus annotation with CellVote — PBMC3k#
This tutorial runs ov.single.CellVote to combine five independent cell type
annotation methods into a single consensus label per cluster, addressing
issue #694 (updated
CellVote to pick the best celltype).
The input adata already has five annotation columns produced by the two
upstream tutorials:
source notebook |
obs columns added |
|---|---|
|
|
|
|
CellVote then takes a per-cluster majority within each of these five columns and asks an LLM to arbitrate the final label using the cluster’s top marker genes as context.
Setup: API key for the arbitration step#
CellVote calls an LLM in the last step to pick between candidate labels.
Any OpenAI-compatible chat endpoint works. Sign up at one of the providers
below, export your key as AGI_API_KEY before launching Jupyter, and
configure provider / base_url / model on the vote() call.
Provider |
Sign up / get a key |
Recommended |
|---|---|---|
DeepSeek (cheap, used here) |
|
|
OpenAI |
|
|
Qwen / Aliyun DashScope |
https://help.aliyun.com/zh/dashscope/developer-reference/get-api-key |
|
Kimi / Moonshot |
|
# in your shell, before launching jupyter:
export AGI_API_KEY=sk-your-actual-key
import os
# `AGI_API_KEY` is the env var omicverse reads for the LLM arbitration step.
# Export it in your shell before launching Jupyter (see the section above).
assert os.environ.get('AGI_API_KEY'), (
'AGI_API_KEY is not set. See the previous cell for provider links.'
)
import anndata as ad
import scanpy as sc
import omicverse as ov
ov.plot_set(font_path='Arial')
🔬 Starting plot initialization...
Using already downloaded Arial font from: /tmp/omicverse_arial.ttf
Registered as: Arial
🧬 Detecting GPU devices…
✅ NVIDIA CUDA GPUs detected: 1
• [CUDA 0] NVIDIA H100 80GB HBM3
Memory: 79.1 GB | Compute: 9.0
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.1.3rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
1) Load the pre-annotated pbmc3k#
data/pbmc3k_5anno.h5ad was produced by running the noref + ref tutorials
end-to-end (celltypist + scsa + gpt4celltype + harmony + scVI) and merging
their outputs on shared cell barcodes.
adata = ad.read_h5ad('data/pbmc3k_5anno.h5ad')
print(adata)
print()
anno_cols = ['celltypist_prediction', 'scsa_prediction', 'gpt4celltype_prediction',
'harmony_prediction', 'scVI_prediction']
for c in anno_cols:
print(f' {c}: {adata.obs[c].nunique()} unique values')
AnnData object with n_obs × n_vars = 2562 × 2000
obs: 'nUMIs', 'mito_perc', 'ribo_perc', 'hb_perc', 'detected_genes', 'cell_complexity', 'n_counts', 'total_counts', 'n_genes', 'n_genes_by_counts', 'pct_counts_mt', 'pct_counts_ribo', 'pct_counts_hb', 'passing_mt', 'passing_nUMIs', 'passing_ngenes', 'predicted_doublet', 'doublet_score', 'scdblfinder_doublet', 'scdblfinder_score', 'leiden', 'celltypist_prediction', 'scsa_prediction', 'gpt4celltype_prediction', 'harmony_prediction', 'scVI_prediction'
var: 'gene_ids', 'mt', 'ribo', 'hb', 'n_cells', 'percent_cells', 'robust', 'highly_variable_features', 'means', 'variances', 'residual_variances', 'highly_variable_rank', 'highly_variable'
uns: 'REFERENCE_MANU', '_ov_provenance', 'history_log', 'hvg', 'leiden', 'log1p', 'neighbors', 'over_clustering', 'pca', 'rank_genes_groups', 'scaled|original|cum_sum_eigenvalues', 'scaled|original|pca_var_ratios', 'status', 'status_args', 'umap'
obsm: 'X_pca', 'X_umap', 'celltypist_decision_matrix', 'celltypist_probability_matrix', 'scaled|original|X_pca'
varm: 'PCs', 'scaled|original|pca_loadings'
layers: 'counts', 'scaled'
obsp: 'connectivities', 'distances'
celltypist_prediction: 11 unique values
scsa_prediction: 7 unique values
gpt4celltype_prediction: 8 unique values
harmony_prediction: 18 unique values
scVI_prediction: 17 unique values
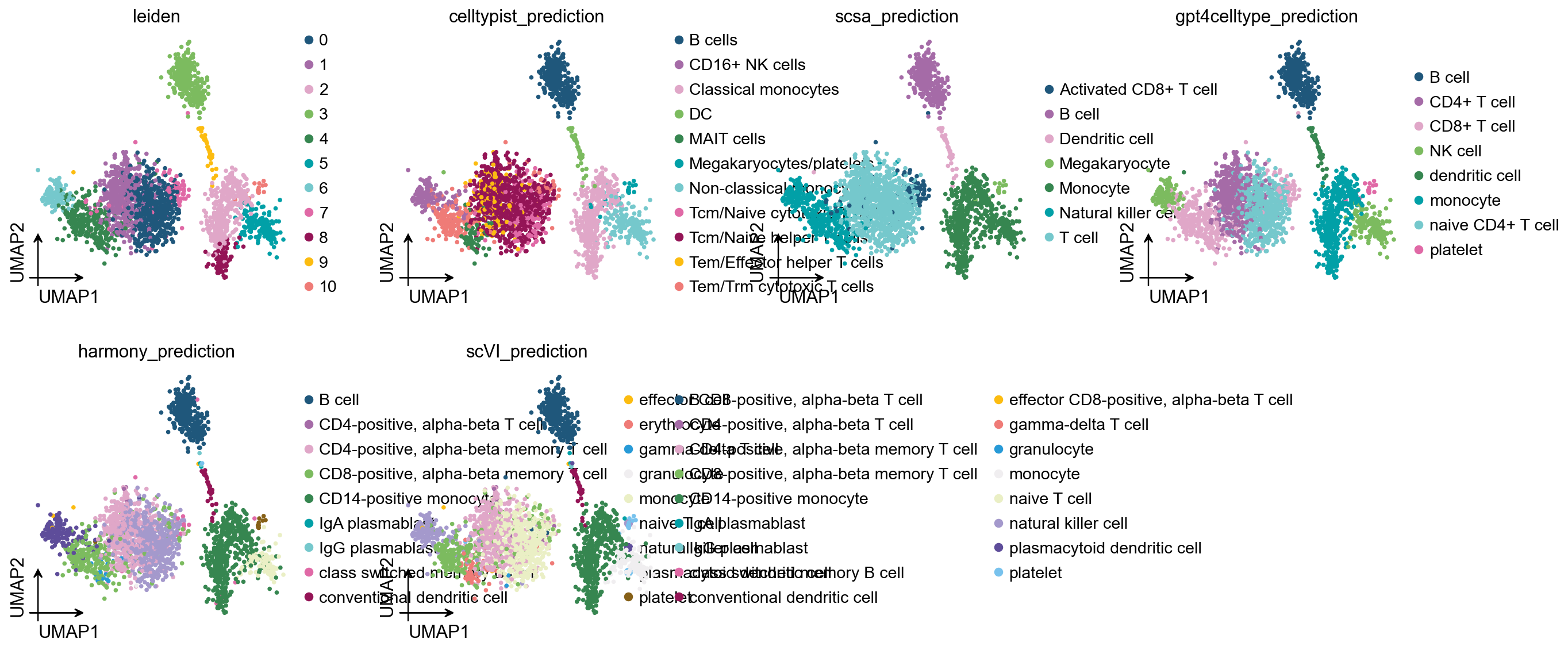
2) Compare the five annotations on UMAP#
ov.pl.umap(
adata,
color=['leiden'] + anno_cols,
frameon='small', palette=ov.pl.sc_color, ncols=4,
)
X_umap converted to UMAP to visualize and saved to adata.obsm['UMAP']
if you want to use X_umap, please set convert=False
3) Compute marker genes per leiden cluster#
CellVote uses the top markers as context when arbitrating between
candidate labels. ov.single.get_celltype_marker returns a
dict[cluster_id -> list[str]].
marker_dict = ov.single.get_celltype_marker(
adata, clustertype='leiden', topgenenumber=10,
foldchange=1.5, pval_cutoff=0.05,
)
for cid, genes in list(marker_dict.items())[:3]:
print(f' leiden={cid}: {genes[:6]}')
...get cell type marker
leiden=0: ['CCR7', 'EPHX2', 'PRKCQ-AS1', 'FHIT', 'LEF1', 'NELL2']
leiden=1: ['CRIP2', 'CD2', 'AQP3', 'CD40LG', 'LTB', 'TRAT1']
leiden=10: ['PF4', 'PPBP', 'SDPR', 'CALM3', 'RGS10', 'GPX1']
4) Run CellVote with all five annotation columns#
CellVote.vote() does two things:
Per cluster, per method — picks the most-frequent label from that method’s annotation column. This yields one candidate per method (so up to 5 candidates per cluster here).
LLM arbitration — sends
{candidates, top markers, species, tissue}to the configured chat model and asks it to choose the most plausible identity. The result is written back toadata.obs[result_key](defaultCellVote_celltype).
cv = ov.single.CellVote(adata)
result_dict = cv.vote(
clusters_key='leiden',
cluster_markers=marker_dict,
celltype_keys=anno_cols, # ← all 5 annotation sources
provider='openai', # use the OpenAI-compatible client
base_url='https://api.deepseek.com/v1',
model='deepseek-chat',
species='human',
organization='PBMC',
)
print()
print('CellVote consensus per cluster:')
for cid, label in sorted(result_dict.items(), key=lambda x: int(x[0]) if str(x[0]).isdigit() else x[0]):
print(f' leiden={cid}: {label}')
CellVote consensus per cluster:
leiden=0: tcm/naive helper t cell
leiden=1: cd4-positive, alpha-beta memory t cell
leiden=2: cd14-positive monocyte
leiden=3: b cell
leiden=4: cd8-positive, alpha-beta memory t cell
leiden=5: non-classical monocyte
leiden=6: cd16+ nk cell
leiden=7: cd8-positive, alpha-beta memory t cell
leiden=8: cd14-positive monocyte
leiden=9: conventional dendritic cell
leiden=10: megakaryocyte/platelet
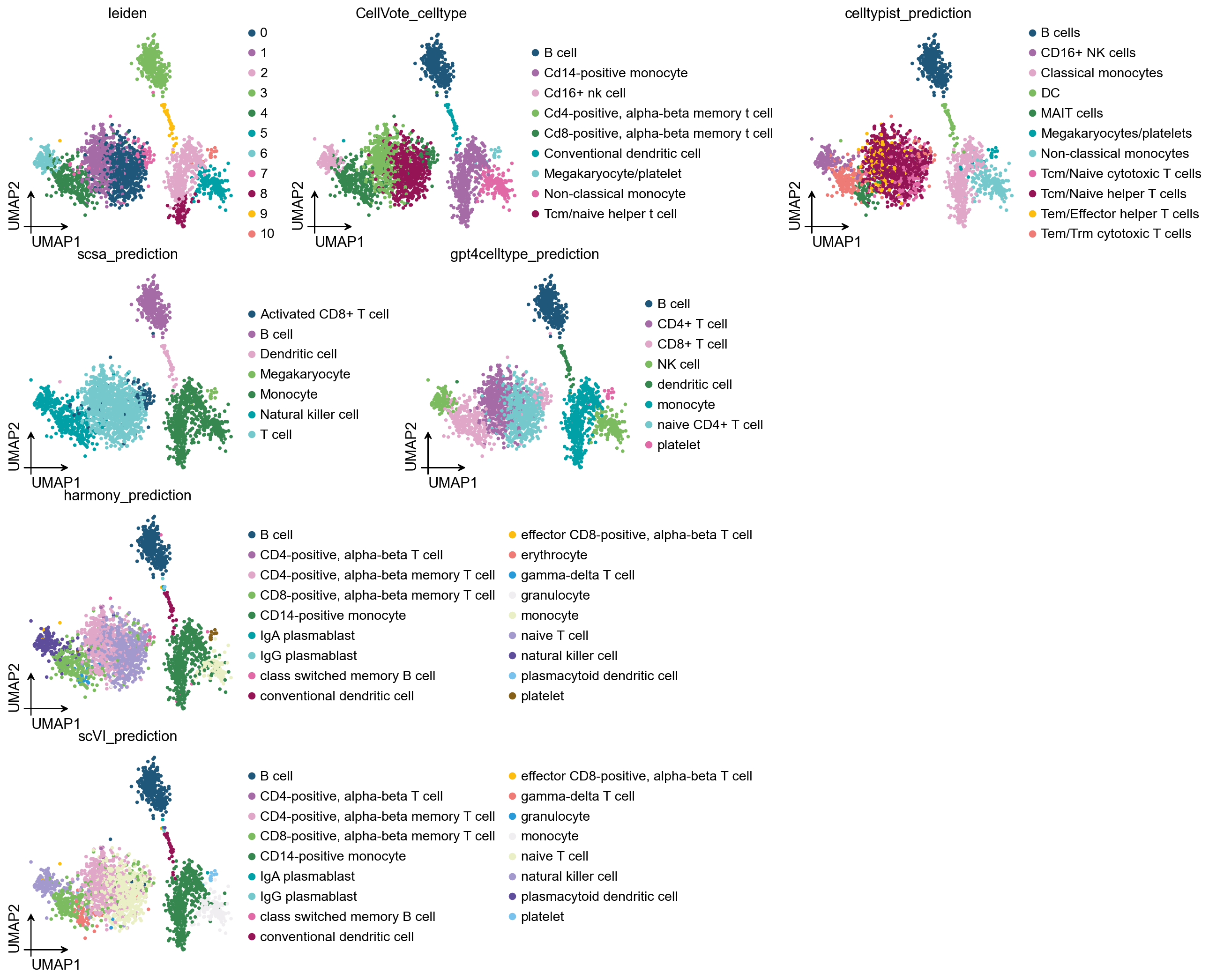
5) Final consensus + per-cluster confidence#
# layout='flow' guarantees no panel-to-panel legend overlap
ov.pl.umap(
adata,
color=['leiden', 'CellVote_celltype'] + anno_cols,
frameon='small', palette=ov.pl.sc_color, ncols=4,
layout='flow',
)
X_umap converted to UMAP to visualize and saved to adata.obsm['UMAP']
if you want to use X_umap, please set convert=False
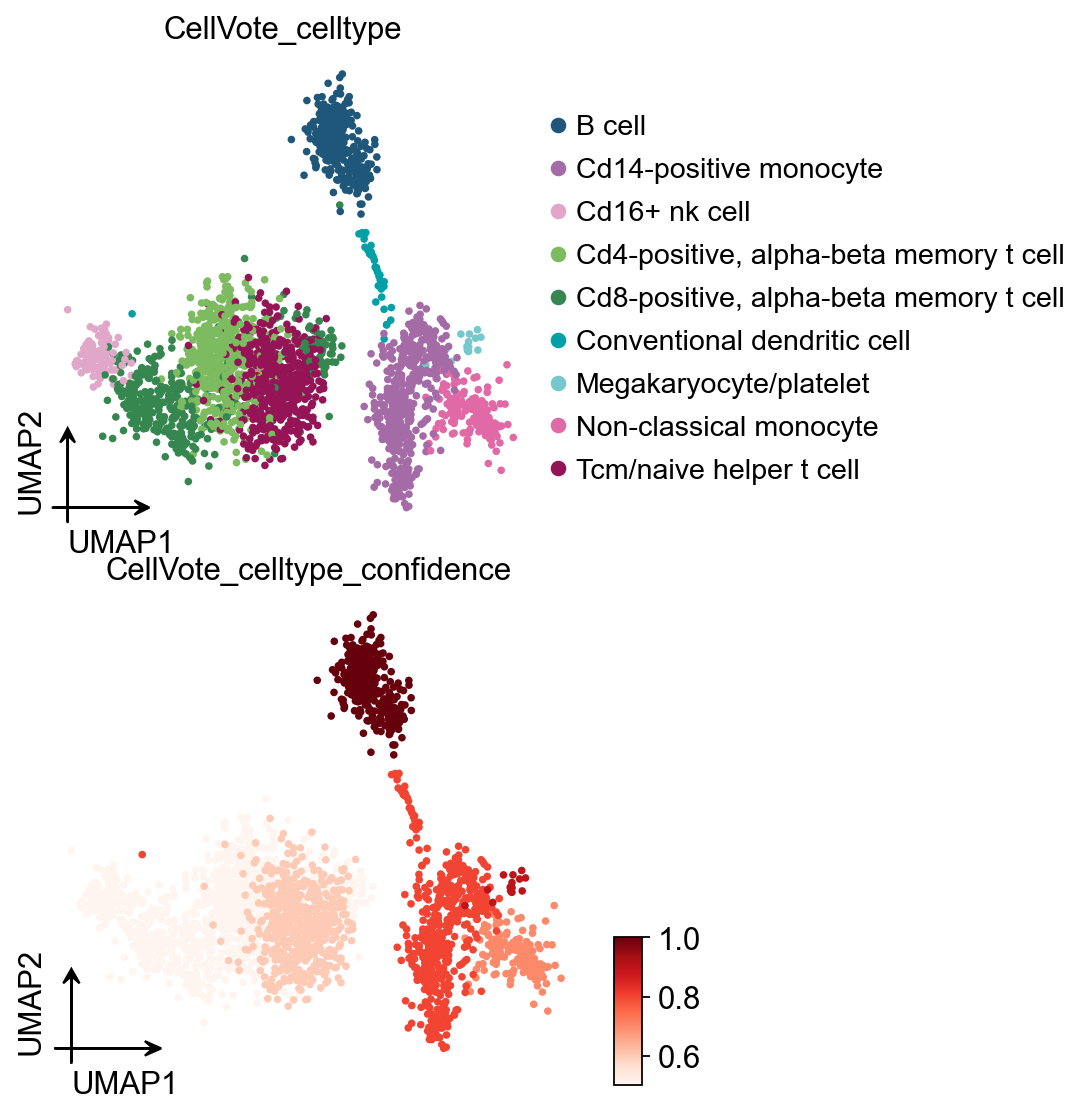
6) CellVote confidence (per-cluster consensus score)#
CellVote.vote() now writes a per-cell confidence column
adata.obs['CellVote_celltype_confidence'] and a per-cluster score
table to adata.uns['CellVote_celltype_score_table'].
The score is built from three metrics on the 5 input methods:
n_unique — distinct labels after normalisation (1..5)
plurality — fraction of methods agreeing with the most common label
vote_agreement — fraction of methods whose label is semantically consistent with the final CellVote pick (token-set Jaccard ≥ 0.34, with synonym + plural normalisation)
confidence —
(plurality + vote_agreement) / 2∈ [0, 1]
Clusters with confidence < 0.6 are the ones most worth a manual look.
import pandas as pd
score_df = pd.DataFrame(adata.uns['CellVote_celltype_score_table'])
score_df = score_df.sort_values('confidence', ascending=False)
score_df
| cluster | n_cells | cellvote_label | n_unique | plurality | vote_agreement | confidence | methods_supporting | |
|---|---|---|---|---|---|---|---|---|
| 3 | 3 | 328 | b cell | 1 | 1.0 | 1.0 | 1.0 | 5/5 |
| 10 | 10 | 16 | megakaryocyte/platelet | 2 | 0.8 | 1.0 | 0.9 | 5/5 |
| 2 | 2 | 376 | cd14-positive monocyte | 2 | 0.6 | 1.0 | 0.8 | 5/5 |
| 8 | 8 | 80 | cd14-positive monocyte | 2 | 0.6 | 1.0 | 0.8 | 5/5 |
| 9 | 9 | 38 | conventional dendritic cell | 2 | 0.6 | 1.0 | 0.8 | 5/5 |
| 5 | 5 | 165 | non-classical monocyte | 3 | 0.6 | 0.8 | 0.7 | 4/5 |
| 0 | 0 | 545 | tcm/naive helper t cell | 4 | 0.4 | 0.8 | 0.6 | 4/5 |
| 1 | 1 | 513 | cd4-positive, alpha-beta memory t cell | 4 | 0.4 | 0.6 | 0.5 | 3/5 |
| 4 | 4 | 283 | cd8-positive, alpha-beta memory t cell | 4 | 0.4 | 0.6 | 0.5 | 3/5 |
| 6 | 6 | 128 | cd16+ nk cell | 3 | 0.6 | 0.4 | 0.5 | 2/5 |
| 7 | 7 | 90 | cd8-positive, alpha-beta memory t cell | 4 | 0.4 | 0.6 | 0.5 | 3/5 |
# Visualise where consensus is strong vs weak
ov.pl.umap(
adata,
color=['CellVote_celltype', 'CellVote_celltype_confidence'],
frameon='small', palette=ov.pl.sc_color, ncols=4,
cmap='Reds',
layout='flow',
)
X_umap converted to UMAP to visualize and saved to adata.obsm['UMAP']
if you want to use X_umap, please set convert=False
7) Per-cluster consensus table (5 methods + final + confidence)#
cols_for_table = anno_cols + ['CellVote_celltype']
summary = (
adata.obs.groupby('leiden', observed=True)[cols_for_table]
.agg(lambda s: s.value_counts().index[0] if len(s) else '')
)
summary['n_cells'] = adata.obs.groupby('leiden', observed=True).size()
summary['confidence'] = pd.DataFrame(
adata.uns['CellVote_celltype_score_table']
).set_index('cluster')['confidence']
summary = summary[['n_cells'] + cols_for_table + ['confidence']]
summary
| n_cells | celltypist_prediction | scsa_prediction | gpt4celltype_prediction | harmony_prediction | scVI_prediction | CellVote_celltype | confidence | |
|---|---|---|---|---|---|---|---|---|
| leiden | ||||||||
| 0 | 545 | Tcm/Naive helper T cells | T cell | naive CD4+ T cell | naive T cell | naive T cell | Tcm/naive helper t cell | 0.6 |
| 1 | 513 | Tcm/Naive helper T cells | T cell | CD4+ T cell | CD4-positive, alpha-beta memory T cell | CD4-positive, alpha-beta memory T cell | Cd4-positive, alpha-beta memory t cell | 0.5 |
| 2 | 376 | Classical monocytes | Monocyte | monocyte | CD14-positive monocyte | CD14-positive monocyte | Cd14-positive monocyte | 0.8 |
| 3 | 328 | B cells | B cell | B cell | B cell | B cell | B cell | 1.0 |
| 4 | 283 | Tem/Trm cytotoxic T cells | Natural killer cell | CD8+ T cell | CD8-positive, alpha-beta memory T cell | CD8-positive, alpha-beta memory T cell | Cd8-positive, alpha-beta memory t cell | 0.5 |
| 5 | 165 | Non-classical monocytes | Monocyte | NK cell | monocyte | monocyte | Non-classical monocyte | 0.7 |
| 6 | 128 | CD16+ NK cells | Natural killer cell | NK cell | natural killer cell | natural killer cell | Cd16+ nk cell | 0.5 |
| 7 | 90 | Tem/Trm cytotoxic T cells | Activated CD8+ T cell | CD8+ T cell | CD8-positive, alpha-beta memory T cell | CD8-positive, alpha-beta memory T cell | Cd8-positive, alpha-beta memory t cell | 0.5 |
| 8 | 80 | Classical monocytes | Monocyte | monocyte | CD14-positive monocyte | CD14-positive monocyte | Cd14-positive monocyte | 0.8 |
| 9 | 38 | DC | Dendritic cell | dendritic cell | conventional dendritic cell | conventional dendritic cell | Conventional dendritic cell | 0.8 |
| 10 | 16 | Megakaryocytes/platelets | Megakaryocyte | platelet | platelet | platelet | Megakaryocyte/platelet | 0.9 |
8) Notes#
Replace
provider/base_url/modelwith any OpenAI-compatible chat endpoint (openai,kimi,qwen,doubao, or a self-hosted model).result_keyoverrides the output column name if you want to keep multiple CellVote runs side-by-side. The confidence column is namedf"{result_key}_confidence"accordingly.cluster_markersis required — the LLM uses gene context to break ties between candidate labels.The confidence score is purely token-level (no extra LLM call); if you want stricter ontology-aware scoring, swap the threshold or run the
cellvote_consensus_score()helper with a customjaccard_threshold.