16S phylogeny: MAFFT → FastTree → Faith PD + UniFrac#
Build a phylogenetic tree from ASV centroids and unlock the tree-aware diversity metrics (Faith’s PD, weighted / unweighted UniFrac) that the plain 16S tutorial couldn’t run.
Pipeline (all real tool wrappers in ov.alignment):
Step |
Tool |
Function |
|---|---|---|
1 |
MAFFT |
|
2 |
FastTree / FastTreeMP |
|
— |
both as one call |
|
Downstream (in ov.micro):
ov.micro.attach_tree(adata, newick=...)— store tree onadata.uns['tree'], prune tovar_namesov.micro.Alpha(adata).run(['faith_pd', ...])— tree-aware alpha diversityov.micro.Beta(adata).run(metric='unweighted_unifrac' / 'weighted_unifrac')ov.micro.Ordinate(adata, dist_key='unweighted_unifrac').pcoa(n=3)
No
$HOMEwrites. All intermediates land under theworkdirpath you pass explicitly.
1. Setup#
from pathlib import Path
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import anndata as ad
import omicverse as ov
ov.plot_set()
# Load the AnnData produced by the basic 16S tutorial
BASE = Path('/scratch/users/steorra/analysis/omicverse_dev/cache/16s')
H5AD = BASE / 'run_mothur_sop' / 'mothur_sop_16s.h5ad'
ASVS = BASE / 'run_mothur_sop' / 'asv' / 'asvs.fasta'
WORK = BASE / 'phylogeny_tutorial'
WORK.mkdir(parents=True, exist_ok=True)
adata = ad.read_h5ad(H5AD)
print('adata:', adata.shape)
print('var has taxonomy cols:', [c for c in adata.var.columns if c in ('phylum','genus')])
🔬 Starting plot initialization...
🧬 Detecting GPU devices…
✅ NVIDIA CUDA GPUs detected: 1
• [CUDA 0] NVIDIA H100 80GB HBM3
Memory: 79.1 GB | Compute: 9.0
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.1.2rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
adata: (20, 598)
var has taxonomy cols: ['phylum', 'genus']
2. Build the phylogenetic tree#
MAFFT aligns all 598 ASV centroids, then FastTree(MP) infers the tree. ~4 seconds on 8 CPU.
tree = ov.alignment.build_phylogeny(
str(ASVS),
workdir=str(WORK),
mafft_threads=8,
fasttree_threads=4,
)
print('aligned:', tree['aligned'])
print('tree :', tree['tree'])
print('newick (first 120 chars):', tree['newick'][:120])
>> /home/users/steorra/miniforge3/envs/omicverse/bin/mafft --thread 8 --auto /scratch/users/steorra/analysis/omicverse_dev/cache/16s/run_mothur_sop/asv/asvs.fasta
>> /home/users/steorra/miniforge3/envs/omicverse/bin/FastTreeMP -nt -gtr -gamma /scratch/users/steorra/analysis/omicverse_dev/cache/16s/phylogeny_tutorial/aligned/aligned.fasta
aligned: /scratch/users/steorra/analysis/omicverse_dev/cache/16s/phylogeny_tutorial/aligned/aligned.fasta
tree : /scratch/users/steorra/analysis/omicverse_dev/cache/16s/phylogeny_tutorial/tree/tree.nwk
newick (first 120 chars): (((((F3D0.1908:0.019502580,(F3D0.682:0.000000006,F3D0.908:0.004693323)0.000:0.000000006)0.957:0.025586231,((F3D0.4522:0.
3. Attach the tree to AnnData#
attach_tree prunes tips to var_names, midpoint-roots the tree (FastTree
emits unrooted), and writes the newick to adata.uns['tree'].
ov.micro.attach_tree(adata, newick=tree['newick'])
print("uns['tree'] len:", len(adata.uns['tree']))
print("tree_tips :", adata.uns['micro']['tree_tips'])
uns['tree'] len: 21046
tree_tips : 598
4. Phylogenetic alpha: Faith PD#
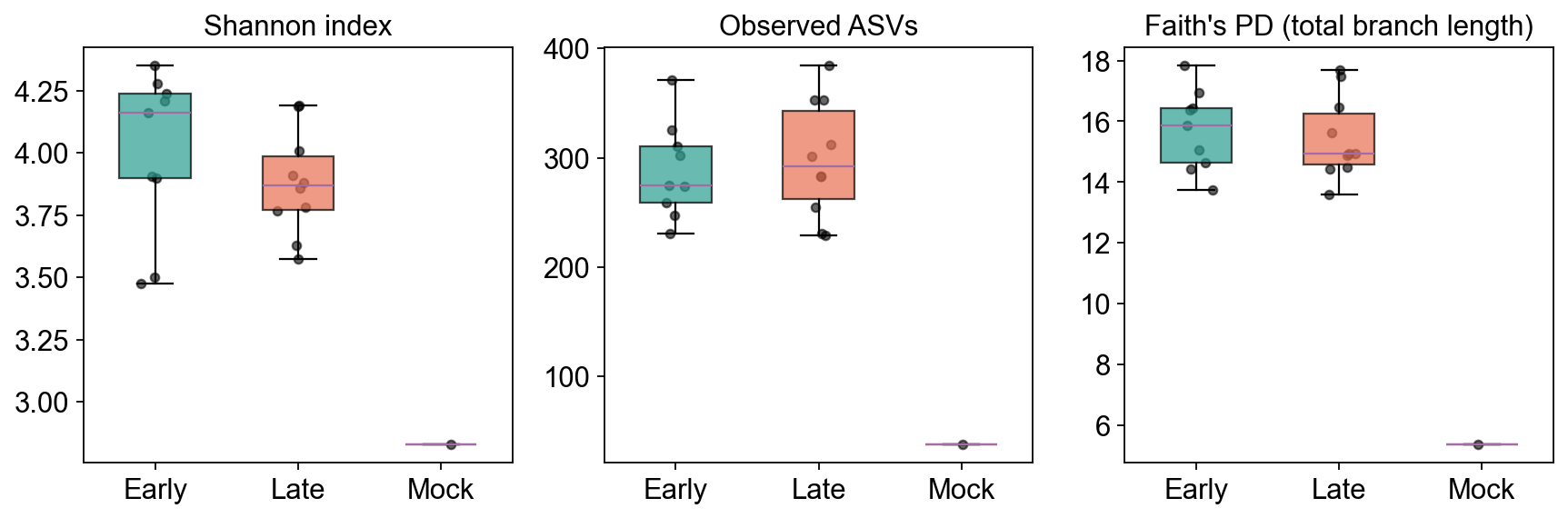
Faith’s Phylogenetic Diversity = the total branch length of the subtree spanned by a sample’s observed ASVs. Unlike Observed / Shannon (which count ASVs agnostically), Faith PD rewards samples whose ASVs are spread across the tree.
import re
# rebuild day + group metadata (not always round-tripped through h5ad)
for name in adata.obs_names:
m = re.match(r'F3D(\d+)', name)
day = int(m.group(1)) if m else -1
group = 'Early' if m and day <= 9 else ('Late' if m else 'Mock')
adata.obs.loc[name, 'day'] = day
adata.obs.loc[name, 'group'] = group
adata.obs['day'] = adata.obs['day'].astype(int)
alpha = ov.micro.Alpha(adata).run(metrics=['shannon', 'observed_otus', 'faith_pd'])
alpha.describe()
shannon observed_otus faith_pd
count 20.000000 20.000000 20.000000
mean 3.880857 280.800000 15.060490
std 0.362040 73.562433 2.608692
min 2.830575 38.000000 5.385063
25% 3.731984 253.000000 14.484808
50% 3.901867 283.000000 15.001241
75% 4.186014 315.250000 16.445646
max 4.348582 384.000000 17.822771
# Compare the three alpha metrics across Early / Late / Mock
palette = {'Early': '#2a9d8f', 'Late': '#e76f51', 'Mock': '#264653'}
groups = ['Early', 'Late', 'Mock']
fig, axes = plt.subplots(1, 3, figsize=(11, 3.8))
for ax, metric, ylabel in zip(
axes,
['shannon', 'observed_otus', 'faith_pd'],
['Shannon index', 'Observed ASVs', "Faith's PD (total branch length)"],
):
data = [adata.obs[adata.obs.group == g][metric].dropna().values for g in groups]
bp = ax.boxplot(data, labels=groups, patch_artist=True, widths=0.5, showfliers=False)
for patch, g in zip(bp['boxes'], groups):
patch.set_facecolor(palette[g]); patch.set_alpha(0.7)
for i, (g, vals) in enumerate(zip(groups, data), start=1):
x = np.random.normal(i, 0.05, size=len(vals))
ax.scatter(x, vals, color='k', alpha=0.6, s=18)
ax.set_title(ylabel)
plt.tight_layout()
plt.show()
5. Phylogenetic beta: UniFrac#
UniFrac = the fraction of branch length unique to one of two samples.
Unweighted: presence/absence only — emphasises rare taxa
Weighted: weighted by abundance — emphasises dominant taxa
Both are stored into adata.obsp.
b = ov.micro.Beta(adata)
b.run(metric='unweighted_unifrac', rarefy=False)
b.run(metric='weighted_unifrac', rarefy=False)
uu = adata.obsp['unweighted_unifrac']
wu = adata.obsp['weighted_unifrac']
triu = np.triu_indices_from(uu, k=1)
print(f"unweighted UniFrac: mean off-diag = {uu[triu].mean():.3f}")
print(f"weighted UniFrac : mean off-diag = {wu[triu].mean():.3f}")
unweighted UniFrac: mean off-diag = 0.362
weighted UniFrac : mean off-diag = 0.222
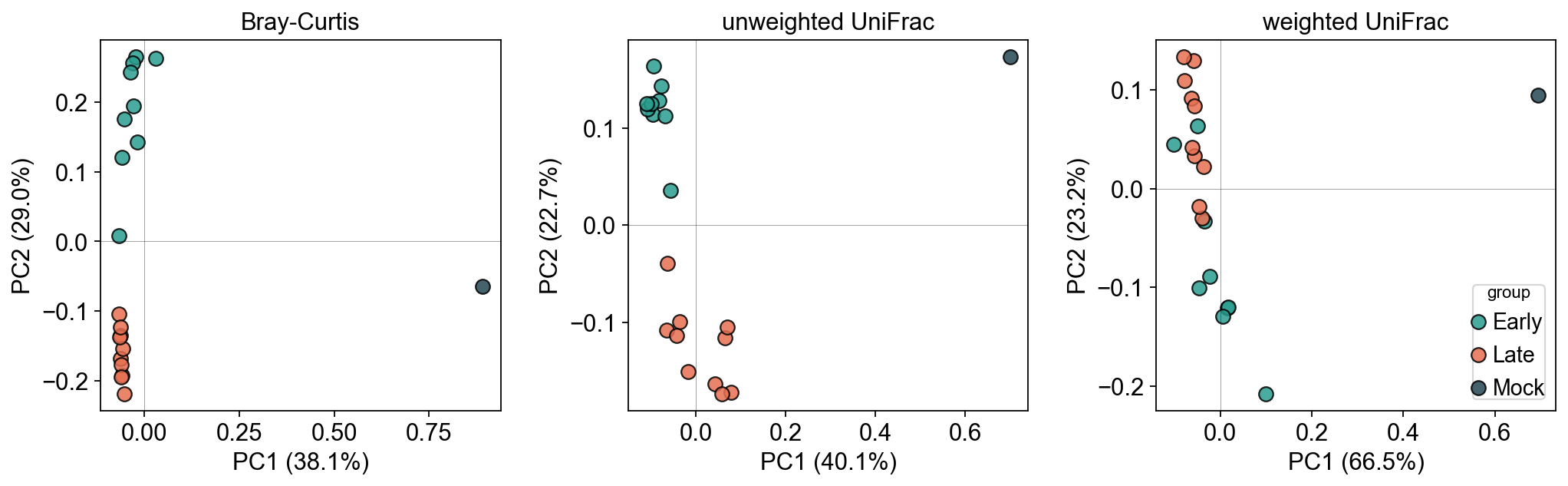
6. Compare ordinations: Bray-Curtis vs UniFrac#
Ordinate each of the three distance matrices with PCoA. If the phylogenetic structure carries additional signal, UniFrac PCoA should separate Early vs Late even more clearly than Bray-Curtis did.
# Bray-Curtis as a baseline
b.run(metric='braycurtis', rarefy=True)
fig, axes = plt.subplots(1, 3, figsize=(13, 4.2))
for ax, dist_key, title in zip(
axes,
['braycurtis', 'unweighted_unifrac', 'weighted_unifrac'],
['Bray-Curtis', 'unweighted UniFrac', 'weighted UniFrac'],
):
ord_ = ov.micro.Ordinate(adata, dist_key=dist_key)
ord_.pcoa(n=2)
pct = ord_.proportion_explained() * 100.0
coords = pd.DataFrame(adata.obsm[f'{dist_key}_pcoa'],
index=adata.obs_names, columns=['PC1','PC2'])
for g in groups:
sub = adata.obs[adata.obs.group == g]
if sub.empty: continue
ax.scatter(coords.loc[sub.index, 'PC1'],
coords.loc[sub.index, 'PC2'],
color=palette[g], s=70, alpha=0.85,
edgecolor='k', label=g)
ax.set_xlabel(f'PC1 ({pct[0]:.1f}%)')
ax.set_ylabel(f'PC2 ({pct[1]:.1f}%)')
ax.set_title(title)
ax.axhline(0, color='k', lw=0.4, alpha=0.4)
ax.axvline(0, color='k', lw=0.4, alpha=0.4)
axes[-1].legend(title='group', loc='best', frameon=True)
plt.tight_layout()
plt.show()
7. Save the tree-enriched AnnData#
out = BASE / 'run_mothur_sop' / 'mothur_sop_16s_tree.h5ad'
adata.write_h5ad(out)
print('saved', out, '-', out.stat().st_size // 1024, 'KB')
saved /scratch/users/steorra/analysis/omicverse_dev/cache/16s/run_mothur_sop/mothur_sop_16s_tree.h5ad - 455 KB
Notes#
Rooting. FastTree produces an unrooted tree;
_tree_objectmidpoint-roots it before passing to scikit-bio’s Faith PD / UniFrac, which require rooted input. Midpoint rooting is neutral wrt outgroup choice and is the canonical choice for ASV trees.SEPP alternative. For a phylogenetically-accurate tree (insertion into a reference backbone), QIIME 2’s SEPP plugin is the gold standard. That’s a follow-up notebook —
ov.alignment.build_phylogenyis the novo-from-ASVs path.Rarefaction. UniFrac is less sensitive to library-size differences than Bray-Curtis, which is why we pass
rarefy=Falseby default here. Passrarefy=Truefor the strictest comparison.