Multi-factor designs — ASCA and linear mixed models#
Most metabolomics studies are not two-group comparisons. A typical
design might be treatment × time with patient IDs (repeated
measures). The two-group metabol.differential test is the wrong tool
here.
ov.metabol.asca(Smilde 2005) — ANOVA-Simultaneous Component Analysis. Decomposes the data into per-factor effect matrices plus pairwise interactions, runs PCA on each, and reports variance-explained + permutation p-value.ov.metabol.mixed_model— per-featurestatsmodels.MixedLMwith a user-defined formula and a random-effect grouping variable (e.g. patient ID).
This tutorial uses a synthetic 2×2 factorial (treatment × time, 6 patients × 4 cells = 24 samples × 25 features) with:
Strong treatment effect on features 0–4
Moderate time effect on features 5–9
Per-patient random intercept
0 — Setup and synthetic factorial#
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
from anndata import AnnData
import omicverse as ov
rng = np.random.default_rng(1)
n_per_cell = 6 # patients
treatments = ['ctrl', 'drug']
times = ['0h', '24h']
n_features = 25
rows, idx = [], []
for t in treatments:
for tm in times:
for k in range(n_per_cell):
rows.append({'treatment': t, 'time': tm, 'patient': f'p{k}'})
idx.append(f'{t}_{tm}_{k}')
obs = pd.DataFrame(rows, index=idx)
n = len(obs)
X = rng.standard_normal((n, n_features)) * 0.3
tmask = (obs['treatment'] == 'drug').to_numpy()
tm_mask = (obs['time'] == '24h').to_numpy()
X[tmask, 0:5] += 2.0 # treatment effect
X[tm_mask, 5:10] += 1.0 # time effect
# Per-patient random intercept (what MixedLM will estimate)
intercepts = rng.standard_normal(n_per_cell) * 1.0
for i, p in enumerate(obs['patient']):
X[i, :] += intercepts[int(p[1:])]
var = pd.DataFrame(index=[f'feat{i}' for i in range(n_features)])
adata = AnnData(X=X, obs=obs, var=var)
adata.shape
(24, 25)
1 — ASCA#
Decompose into treatment, time, treatment:time interaction and
residual. Use 500 permutations for significance testing.
res = ov.metabol.asca(
adata,
factors=['treatment', 'time'],
include_interactions=True,
n_components=2,
n_permutations=500,
seed=0,
)
res.summary()
effect ss df variance_explained p_value
0 treatment 115.937051 1.0 0.160197 0.023952
1 time 31.687051 1.0 0.043784 0.365269
2 treatment:time 1.149195 1.0 0.001588 1.000000
3 residual 574.942287 NaN 0.794431 NaN
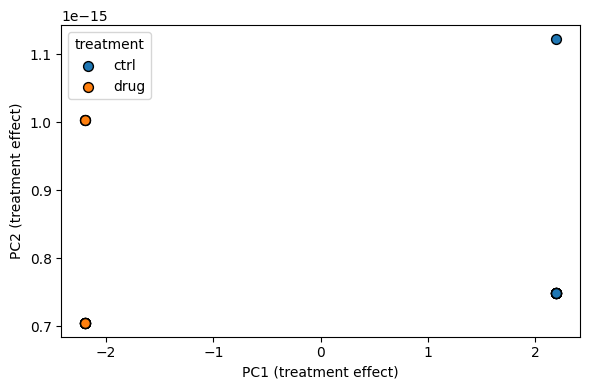
Scores plot for the treatment effect#
The scores matrix of the treatment effect places each sample on a
2-D projection of the factor’s subspace. Samples of the same level
should cluster tightly.
scores = res.scores_frame('treatment')
fig, ax = plt.subplots(figsize=(6, 4))
for lvl, colour in zip(['ctrl', 'drug'], ['C0', 'C3']):
mask = (adata.obs['treatment'] == lvl).to_numpy()
ax.scatter(scores.loc[mask, 'PC1'], scores.loc[mask, 'PC2'],
label=lvl, s=50, edgecolor='k')
ax.set_xlabel('PC1 (treatment effect)')
ax.set_ylabel('PC2 (treatment effect)')
ax.legend(title='treatment')
fig.tight_layout()
plt.show()
Loadings — which features drive the treatment effect?#
load = res.loadings_frame('treatment')
top_treat = load['PC1'].abs().sort_values(ascending=False).head(8)
print('Top treatment-driving features (|PC1 loading|):')
print(top_treat)
Top treatment-driving features (|PC1 loading|):
feat3 0.467249
feat2 0.462797
feat1 0.446272
feat4 0.425777
feat0 0.408436
feat12 0.067782
feat18 0.050729
feat24 0.049333
Name: PC1, dtype: float64
2 — Linear mixed model#
For a specific per-feature effect size + p-value, fit MixedLM with
treatment + time as fixed effects and patient as the random
intercept. Ask for the treatment[T.drug] contrast in short format
to get the same schema as metabol.differential.
tbl = ov.metabol.mixed_model(
adata,
formula='treatment + time',
groups='patient',
term='treatment[T.drug]',
)
tbl.sort_values('pvalue').head(10)
coef se stat pvalue padj
feature
feat3 2.053924 0.103741 19.798582 3.061911e-87 7.654778e-86
feat2 2.034350 0.110201 18.460316 4.308156e-76 5.385195e-75
feat4 1.871621 0.113259 16.525132 2.419114e-61 2.015928e-60
feat0 1.795393 0.108829 16.497441 3.827711e-61 2.392320e-60
feat1 1.961713 0.144438 13.581672 5.143839e-42 2.571919e-41
feat11 0.199102 0.072605 2.742262 6.101756e-03 2.542398e-02
feat12 0.297956 0.128181 2.324500 2.009873e-02 7.178117e-02
feat7 0.213406 0.101708 2.098218 3.588586e-02 1.121433e-01
feat6 0.200778 0.099124 2.025526 4.281340e-02 1.189261e-01
feat18 0.222992 0.121534 1.834820 6.653242e-02 1.663311e-01
The top features should match the planted-signal block (0–4).
FDR correction is applied automatically (padj column).
n_sig = (tbl['padj'] < 0.05).sum()
print(f'{n_sig} features with padj < 0.05 for treatment[T.drug]')
6 features with padj < 0.05 for treatment[T.drug]
3 — MEBA — time-series Hotelling T²#
mixed_model gives you per-feature coefficients for the fixed-effect
design. MEBA (MetaboAnalyst’s time-series module) asks a different
question: does the entire time-course vector differ between
groups? — a multivariate test over time points, not separate tests
at each point.
Requires a balanced design: every subject observed at every time
point. We build a fresh synthetic 2 groups × 4 time points × 8
patients so the API is obvious. On features 0–4 the drug group
gains over time (group × time interaction); features 5–9 have a
time-only trend with no group difference.
rng = np.random.default_rng(2)
n_subj = 8
n_times = 4
group_ids = ['A']*4 + ['B']*4
rows, idx = [], []
for si, s in enumerate([f'p{i}' for i in range(n_subj)]):
for t in range(n_times):
rows.append({'subject': s, 'group': group_ids[si], 'time': f't{t}'})
idx.append(f'{s}_t{t}')
obs_ts = pd.DataFrame(rows, index=idx)
n = len(obs_ts)
X_ts = rng.standard_normal((n, 15)) * 0.3
# Group × time interaction on features 0..4: drug (B) rises over time
for t in range(n_times):
tmask = (obs_ts['time'] == f't{t}').to_numpy()
gmask = (obs_ts['group'] == 'B').to_numpy()
X_ts[tmask & gmask, 0:5] += t * 2.0
# Time-only main effect on features 5..9 — same pattern in both groups
for t in range(n_times):
tmask = (obs_ts['time'] == f't{t}').to_numpy()
X_ts[tmask, 5:10] += t * 1.0
from anndata import AnnData as _AnnData
adata_ts = _AnnData(X=X_ts, obs=obs_ts,
var=pd.DataFrame(index=[f'f{i}' for i in range(15)]))
meba_tbl = ov.metabol.meba(adata_ts, group_col='group',
time_col='time', subject_col='subject')
meba_tbl.sort_values('pvalue').head(10)
T2 F df1 df2 pvalue padj n_a n_b k
f4 8691.685229 1086.460654 4 3 0.000045 0.000679 4 4 4
f0 3909.991043 488.748880 4 3 0.000150 0.001123 4 4 4
f3 2244.084114 280.510514 4 3 0.000344 0.001458 4 4 4
f1 2066.627498 258.328437 4 3 0.000389 0.001458 4 4 4
f2 1569.142040 196.142755 4 3 0.000586 0.001759 4 4 4
f8 140.185920 17.523240 4 3 0.020276 0.050690 4 4 4
f9 39.705078 4.963135 4 3 0.109544 0.234738 4 4 4
f6 12.705891 1.588236 4 3 0.366748 0.667223 4 4 4
f10 11.440460 1.430058 4 3 0.400334 0.667223 4 4 4
f7 8.689745 1.086218 4 3 0.492666 0.738999 4 4 4
MEBA should rank the group×time features (0–4) above the time-only block (5–9) because the shape of the trajectory differs only for the former.
assert meba_tbl.iloc[:5]['F'].mean() > meba_tbl.iloc[5:10]['F'].mean(), \
'unexpected — planted signal not recovered'
print(f"Mean F on interaction block (f0..f4) : {meba_tbl.iloc[:5]['F'].mean():.1f}")
print(f"Mean F on time-only block (f5..f9) : {meba_tbl.iloc[5:10]['F'].mean():.1f}")
print(f"{(meba_tbl['padj'] < 0.05).sum()} features significant at padj < 0.05")
Mean F on interaction block (f0..f4) : 462.0
Mean F on time-only block (f5..f9) : 5.2
5 features significant at padj < 0.05
Takeaways#
asca— global structure; answers which factor explains overall variance?mixed_model— per-feature effect size + p; answers which metabolites change with treatment while respecting patient identity?
Use them together: ASCA to choose which factor is worth testing, MixedLM to get publication-ready per-feature statistics.