Cell-cell communication analysis with LIANA+#
Overview#
This tutorial is built around the LIANA+ rank_aggregate workflow and routes the results into the OmicVerse ccc_* visualization stack.
ov.pl.ccc_heatmapov.pl.ccc_network_plotov.pl.ccc_stat_plot
The point of this structure is to establish, up front, which kinds of biological questions each view answers, and then move into concrete plotting examples.
This notebook keeps two complementary workflows:
Main workflow: pass the original
adatadirectly intoccc_*Optional workflow: explicitly extract
comm_adatafor inspection and comparison examples
Method background#
According to the LIANA documentation and the LIANA+ paper, LIANA+ is a framework for cell-cell communication analysis that harmonizes ligand-receptor resources and multiple scoring strategies. In this notebook we focus on rank_aggregate, which is a strong default because it:
defines sender and receiver groups from cell labels
scores ligand-receptor interactions from normalized expression patterns
aggregates evidence across methods into a consensus ranking
returns a standardized result table that is easy to route into OmicVerse plotting functions
This consensus-style setup is helpful when prioritizing candidate communication axes before pathway-level or network-level interpretation.
Why use pbmc68k_reduced here?#
pbmc68k_reduced is lightweight, already contains UMAP coordinates, and provides a moderate number of immune cell groups. That makes it suitable for demonstrating the LIANA+ inference workflow and the downstream communication plots in one notebook.
import numpy as np
import pandas as pd
import scanpy as sc
import warnings
warnings.filterwarnings("ignore", category=FutureWarning)
import omicverse as ov
ov.plot_set(font_path='Arial')
%reload_ext autoreload
%autoreload 2
🔬 Starting plot initialization...
Using already downloaded Arial font from: /var/folders/rv/3jnfbs0d6r7d0c5bfj7ft5k00000gn/T/omicverse_arial.ttf
Registered as: Arial
🧬 Detecting GPU devices…
✅ Apple Silicon MPS detected
• [MPS] Apple Silicon GPU - Metal Performance Shaders available
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.1.3rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
1. Load the example dataset#
LIANA’s official tutorials also use an AnnData workflow. Here we use pbmc68k_reduced and adata.obs['bulk_labels'] as the communication grouping.
This dataset is a good tutorial choice because:
it is lightweight enough to demonstrate many
ccc_*views end to endit already contains UMAP coordinates for layout checks and embedding-based network plots
the number of cell types is large enough to expose real facet and matrix structure without making every plot collapse
adata = sc.datasets.pbmc68k_reduced()
adata
AnnData object with n_obs × n_vars = 700 × 765
obs: 'bulk_labels', 'n_genes', 'percent_mito', 'n_counts', 'S_score', 'G2M_score', 'phase', 'louvain'
var: 'n_counts', 'means', 'dispersions', 'dispersions_norm', 'highly_variable'
uns: 'bulk_labels_colors', 'louvain', 'louvain_colors', 'neighbors', 'pca', 'rank_genes_groups'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
obsp: 'distances', 'connectivities'
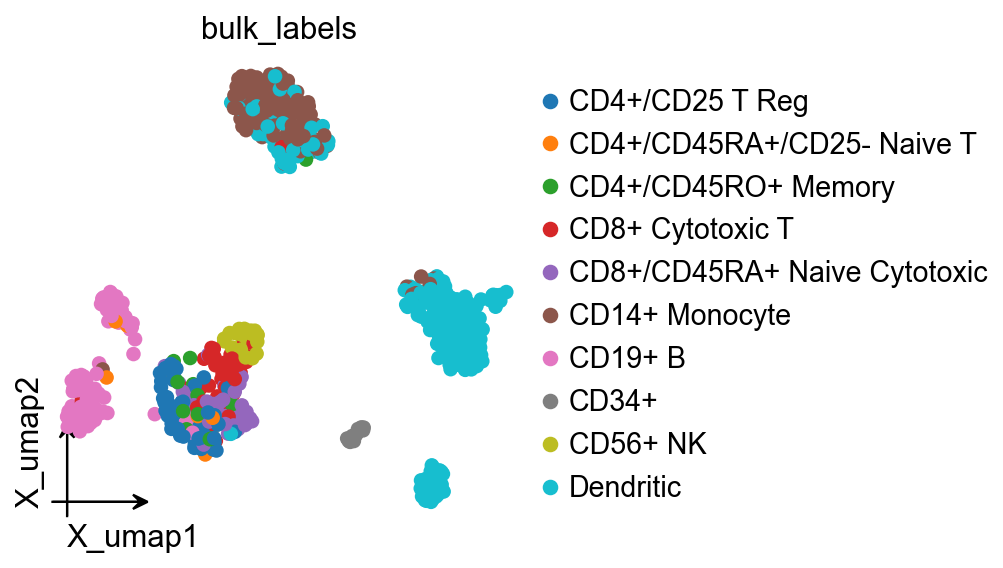
ov.pl.embedding(
adata,
basis='X_umap',
color='bulk_labels',
frameon='small'
)
2. Run LIANA+ rank aggregation#
We use ov.single.run_liana(...) as a wrapper around liana.mt.rank_aggregate and store the result in adata.uns['liana_res'].
Why method='rank_aggregate' is the default here:
the aggregated consensus score is the most natural input for the unified OmicVerse
ccc_*visualizationsit compresses evidence from multiple CCC methods into a single ranking, which is a clear default tutorial entry point
it keeps the result table consistent for downstream pathway-level aggregation and comparison plots
ov.single.run_liana(
adata,
groupby='bulk_labels',
method='rank_aggregate',
resource_name='consensus',
key_added='liana_res',
inplace=True,
)
adata.uns['liana_res'].head()
source target ligand_complex receptor_complex \
1209 Dendritic CD4+/CD45RO+ Memory HLA-DRA CD4
1188 Dendritic CD4+/CD45RA+/CD25- Naive T HLA-DRA CD4
1210 Dendritic CD4+/CD45RO+ Memory HLA-DRB1 CD4
1205 Dendritic CD4+/CD45RO+ Memory HLA-DPB1 CD4
1189 Dendritic CD4+/CD45RA+/CD25- Naive T HLA-DRB1 CD4
lr_means cellphone_pvals expr_prod scaled_weight lr_logfc \
1209 2.575263 0.0 2.780884 0.723815 1.431302
1188 2.566905 0.0 2.705027 0.709428 1.332656
1210 2.415010 0.0 2.584465 0.712731 1.331341
1205 2.367473 0.0 2.526199 0.731297 1.447014
1189 2.406652 0.0 2.513965 0.698344 1.232695
spec_weight lrscore specificity_rank magnitude_rank
1209 0.065077 0.736772 0.001137 0.000653
1188 0.063302 0.734081 0.001137 0.000911
1210 0.060203 0.729607 0.001137 0.001211
1205 0.068953 0.727352 0.001137 0.001377
1189 0.058561 0.726870 0.001137 0.001741
3. Optional: extract the standardized communication AnnData#
Most ccc_* calls no longer require users to manually build comm_adata, because the plotting functions can auto-detect adata.uns['liana_res'].
We still extract it explicitly once for three reasons:
to inspect how LIANA columns are mapped into standardized communication score / p-value layers
to confirm that pathway / classification annotations are available
to simplify the later synthetic comparison and differential-network examples
comm_adata = ov.single.to_comm_adata(
adata,
result_uns_key='liana_res',
score_key='specificity_rank',
pvalue_key='specificity_rank',
classification_reference='cellchat',
classification_fallback='family',
)
comm_adata
AnnData object with n_obs × n_vars = 100 × 42
obs: 'sender', 'receiver', 'cell_type_pair'
var: 'interacting_pair', 'pair_lr', 'interaction_name', 'interaction_name_2', 'classification', 'pathway_name', 'signaling', 'gene_a', 'gene_b', 'ligand', 'receptor', 'annotation_strategy', 'classification_source'
uns: 'liana_score_key', 'liana_pvalue_key', 'liana_uns_key', 'liana_sample_key', 'liana_classification_reference', 'liana_classification_fallback', 'liana_classification_source_counts'
layers: 'means', 'pvalues', 'lr_means', 'cellphone_pvals', 'expr_prod', 'scaled_weight', 'lr_logfc', 'spec_weight', 'lrscore', 'specificity_rank', 'magnitude_rank'
comm_adata.var[['classification', 'classification_source']].value_counts().head()
classification classification_source
MHC-II reference:cellchat_human 10
family 8
ECM/Adhesion family 6
Unclassified unclassified 5
TNF family 3
Name: count, dtype: int64
focus_pathways = [
value
for value in comm_adata.var['classification'].dropna().astype(str).unique().tolist()
if value not in {'Unclassified', 'nan'}
]
focus_pathway = focus_pathways[0] if focus_pathways else 'Unclassified'
focus_pair_lr = comm_adata.var['interacting_pair'].astype(str).iloc[10]
ligand_series = comm_adata.var['ligand'].astype(str)
focus_ligand = ligand_series[ligand_series != ''].iloc[0]
color_dict = dict(
zip(
sorted(adata.obs['bulk_labels'].astype(str).unique()),
ov.pl.sc_color[: adata.obs['bulk_labels'].nunique()],
)
)
umap_df = pd.DataFrame(
adata.obsm['X_umap'][:, :2],
columns=['x', 'y'],
index=adata.obs_names,
)
umap_df['cell_type'] = adata.obs['bulk_labels'].astype(str).values
node_positions = umap_df.groupby('cell_type', observed=True)[['x', 'y']].median()
embedding_points = umap_df.reset_index(drop=True)
comm_adata.uns['node_positions'] = node_positions
comm_adata.uns['embedding_points'] = embedding_points
comm_adata.uns['embedding_axes'] = ('UMAP_1', 'UMAP_2')
comparison_comm = comm_adata.copy()
comparison_comm.layers['means'] = np.asarray(comm_adata.layers['means']).copy() * 0.85
comparison_comm.layers['pvalues'] = np.clip(
np.asarray(comm_adata.layers['pvalues']).copy() * 1.1,
0.0,
1.0,
)
focus_pathway, focus_pair_lr, focus_ligand
('ANNEXIN', 'HLA-DPA1_CD4', 'ANXA1')
1. ov.pl.ccc_heatmap#
This group of plots mainly answers three questions:
which sender / receiver / interaction combinations dominate
which signaling families become most active after pathway-level aggregation
which dense matrix views are better suited for compressed summaries
For LIANA, the first views worth mastering are:
plot_type='dot': the clearest source-target-interaction decompositionplot_type='tile': split ligand-side and receptor-side summariesplot_type='heatmap': pathway/family-level aggregation
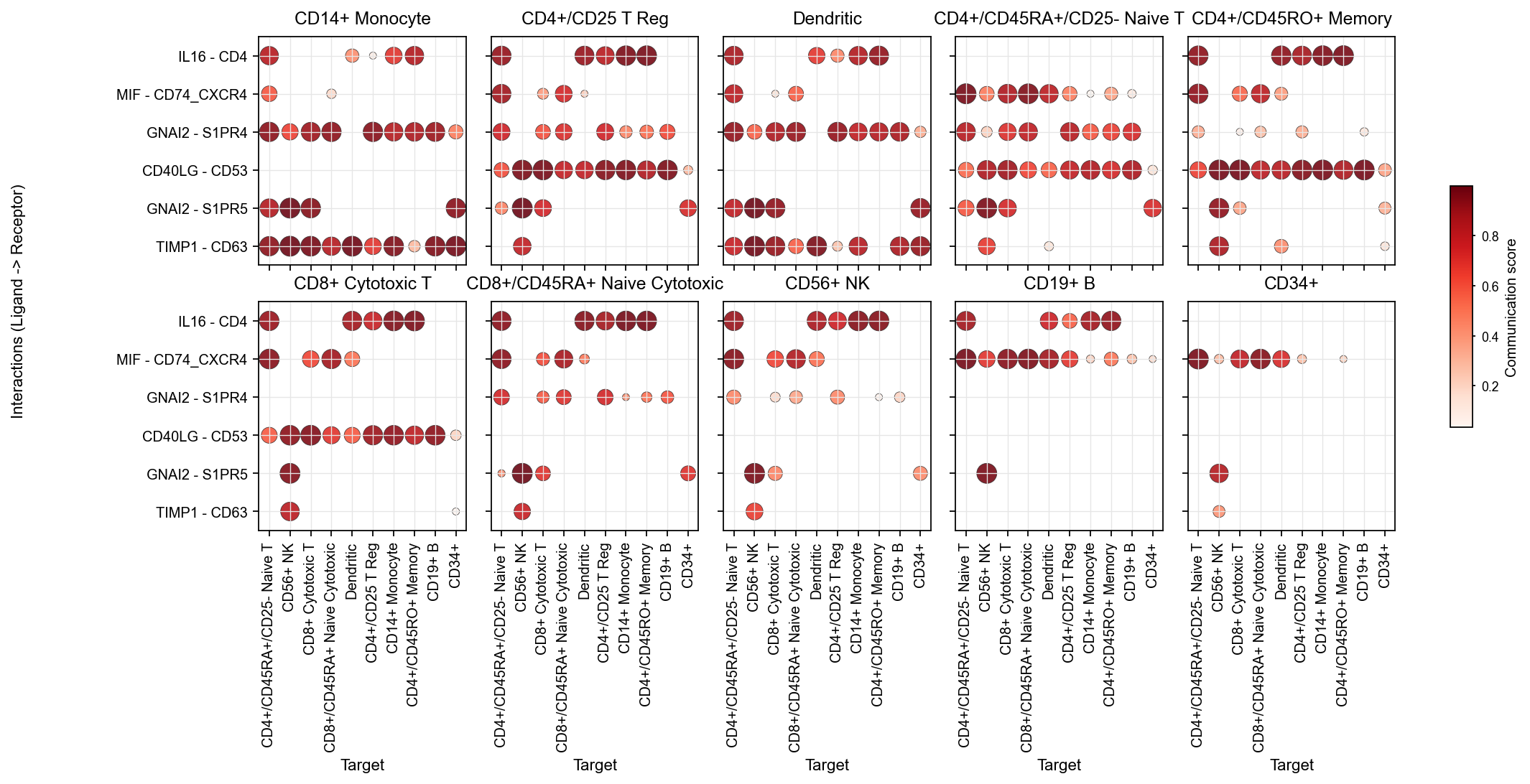
1.1 The main LIANA dot view#
This should be the default entry point for interaction-level inspection.
Facets: source
x-axis: target
y-axis: interaction
color: communication score
It is the clearest way to inspect sender, target, and interaction simultaneously.
fig, ax = ov.pl.ccc_heatmap(
adata,
plot_type='dot',
display_by='interaction',
score_key='specificity_rank',
pvalue_key='specificity_rank',
classification_reference='cellchat',
classification_fallback='family',
top_n=6,
pvalue_threshold=0.05,
color_by='score',
ncols=5,
figsize=(16, 5),
show=False,
)
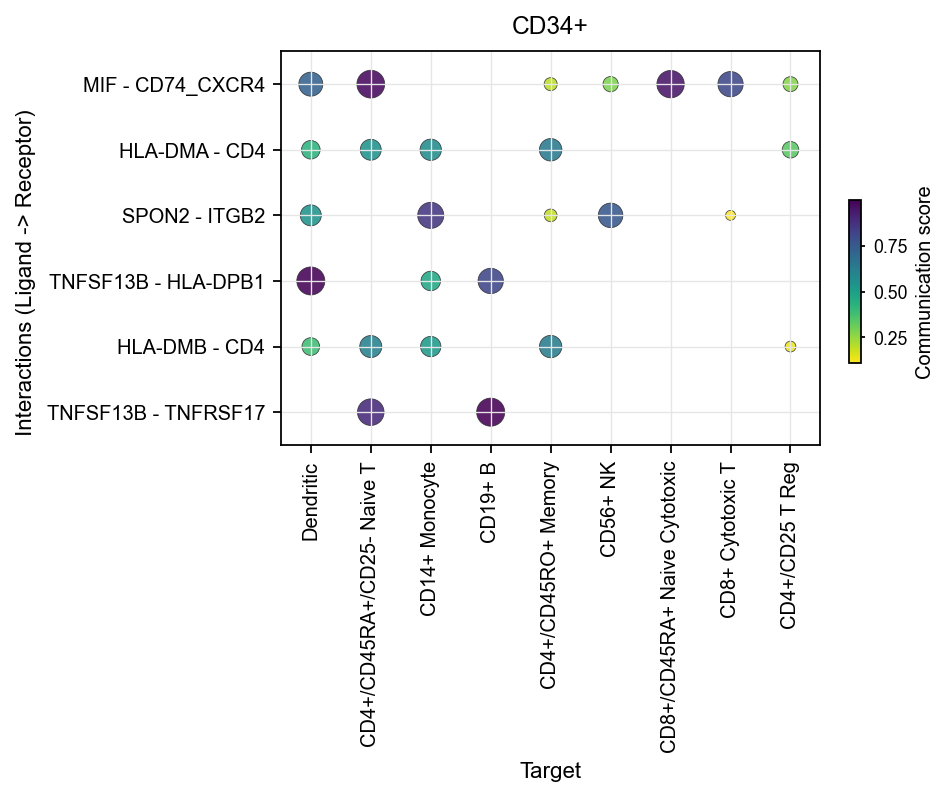
1.2 Sender-focused dot plots#
Once you already know which sender you care about, use sender_use to narrow the view.
This is useful for questions such as:
which targets a given sender primarily communicates with
which interactions dominate that sender
Here, we use the extracted comm_adata using ov. single. to_commaddrata for plotting, taking CD34+ as an example:
fig, ax = ov.pl.ccc_heatmap(
comm_adata,
plot_type='dot',
display_by='interaction',
score_key='specificity_rank',
pvalue_key='specificity_rank',
sender_use='CD34+',
top_n=6,
pvalue_threshold=0.05,
color_by='score',
cmap='viridis_r',
figsize=(6, 3),
show=False,
)
Likewise, you can focus the plot with receiver_use.
fig, ax = ov.pl.ccc_heatmap(
adata,
plot_type='dot',
display_by='interaction',
score_key='specificity_rank',
pvalue_key='specificity_rank',
receiver_use='CD34+',
top_n=5,
pvalue_threshold=0.05,
color_by='score',
cmap='viridis_r',
figsize=(16, 3),
show=False,
)
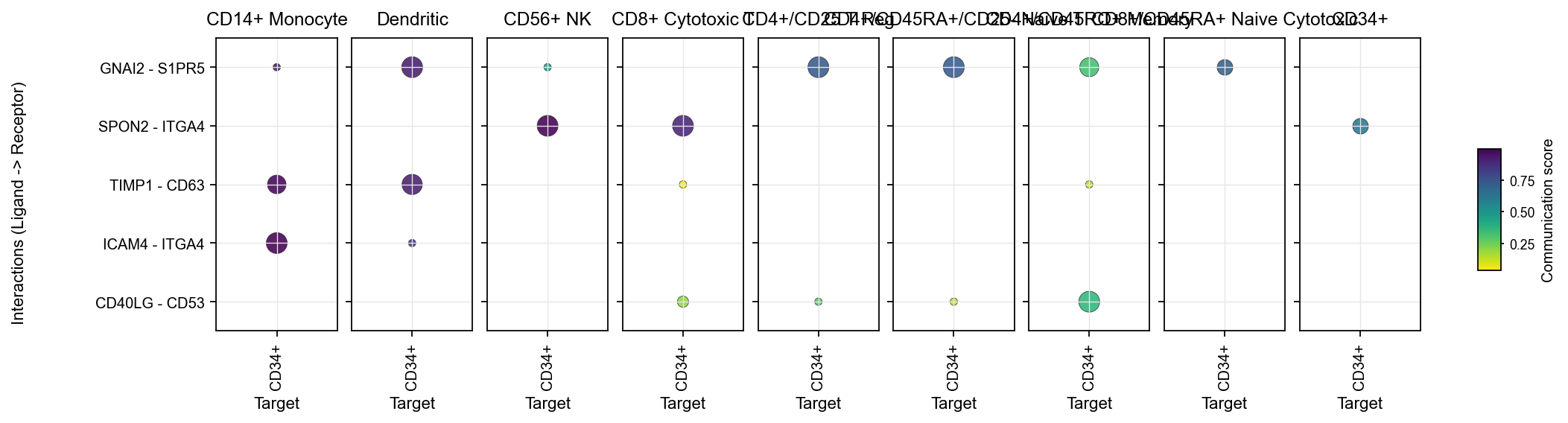
1.3 Tile plots#
tile is useful when you want to look at the same interaction set from the ligand and receptor sides separately.
Left heatmap:
Source ligandRight heatmap:
Target receptor
fig, ax = ov.pl.ccc_heatmap(
adata,
plot_type='tile',
display_by='interaction',
score_key='specificity_rank',
pvalue_key='specificity_rank',
top_n=10,
pvalue_threshold=0.05,
cmap='viridis',
figsize=(8, 3),
show=False,
)
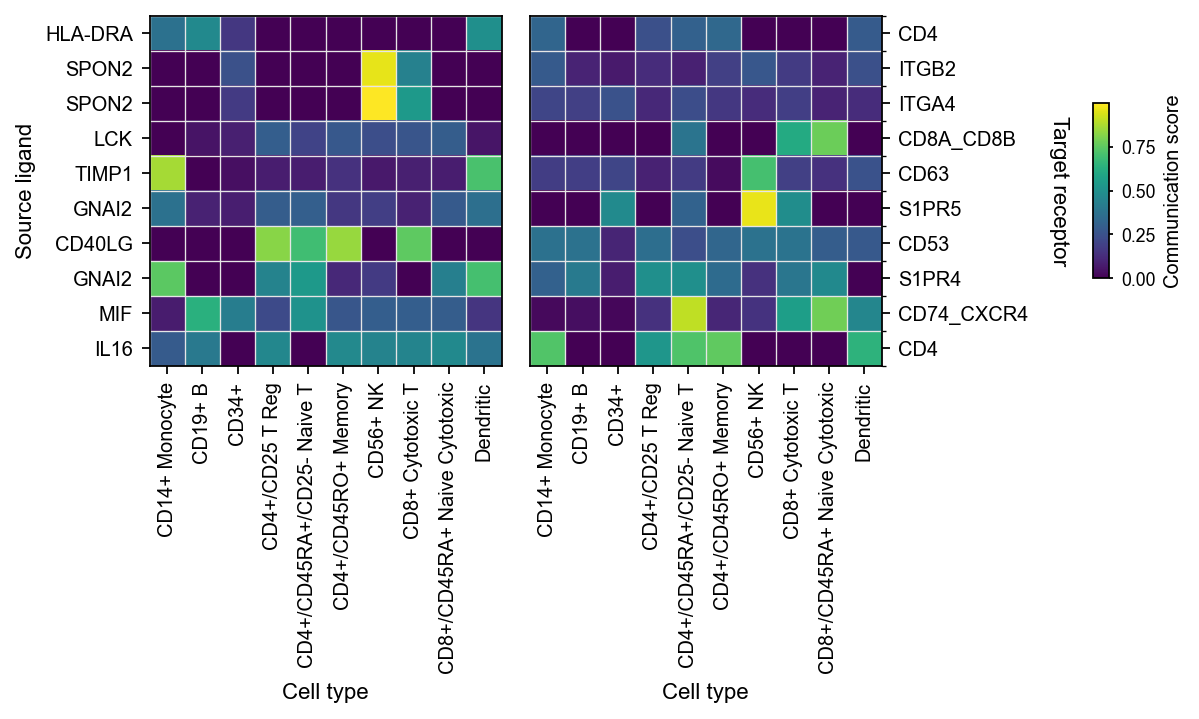
1.4 Pathway aggregation heatmap#
When the question shifts from “which interaction is strongest” to “which signaling family is most active”, the correct view is an aggregation-level heatmap.
This is where classification_reference='cellchat' and classification_fallback='family' really matter, because they determine how ligand-receptor pairs are assigned into pathway/family groups.
fig, ax = ov.pl.ccc_heatmap(
adata,
plot_type='heatmap',
display_by='aggregation',
score_key='specificity_rank',
pvalue_key='specificity_rank',
classification_reference='cellchat',
classification_fallback='family',
top_n=12,
cmap='plasma_r',
show=False,
)
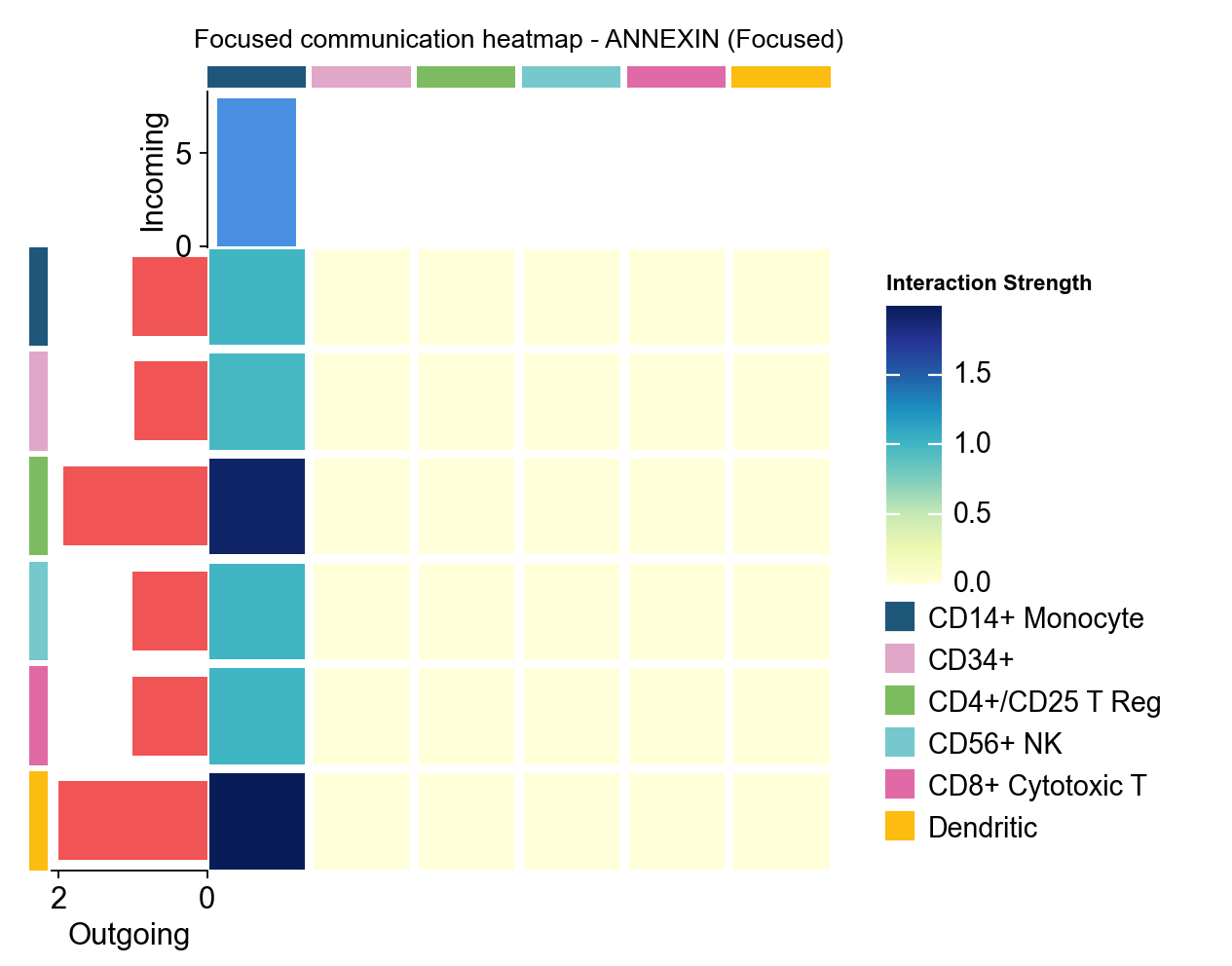
1.5 Focused heatmap#
focused_heatmap is used to pull out one pathway and inspect its sender/receiver structure in isolation.
fig, ax = ov.pl.ccc_heatmap(
adata,
plot_type='focused_heatmap',
signaling=[focus_pathway],
min_interaction_threshold=0.0,
cmap='YlGnBu',
figsize=(4, 3),
show=False,
)
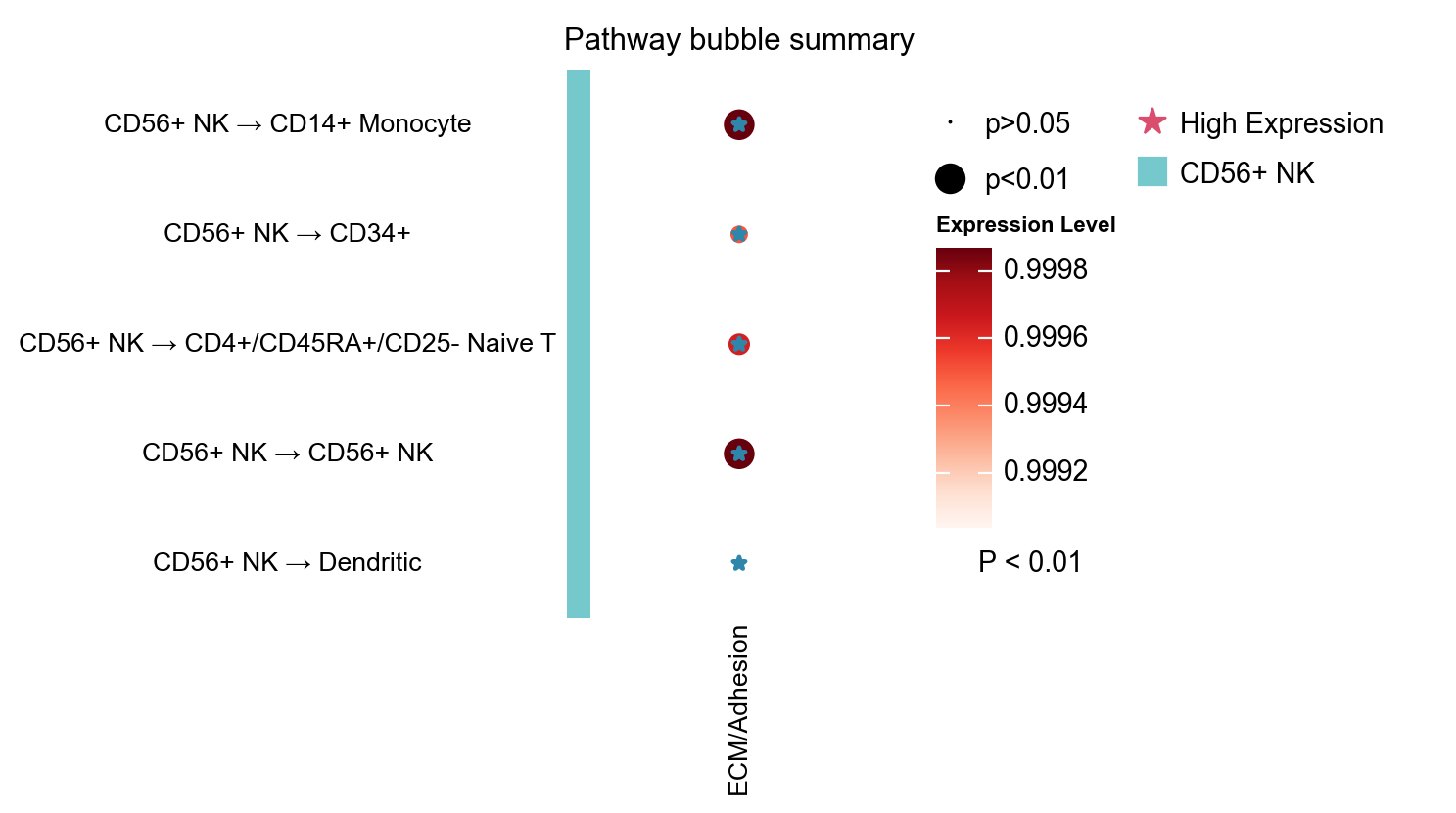
1.6 Pathway bubble, role heatmap, and role network#
These are pathway-level summary views:
pathway_bubble: compact matrix of pathway vs cell-pair activityrole_heatmap: incoming / outgoing summary by cell typerole_network: pathway role relationships organized as a network
fig, ax = ov.pl.ccc_heatmap(
adata,
plot_type='pathway_bubble',
signaling=['ECM/Adhesion'],
top_n=5,
figsize=(3, 5),
show=False,
)
📊 Visualization statistics:
- Number of significant interactions: 5
- Number of cell type pairs: 5
- Signaling pathways: 1
- Data scaling: None (raw expression values)
- Color bars: sender
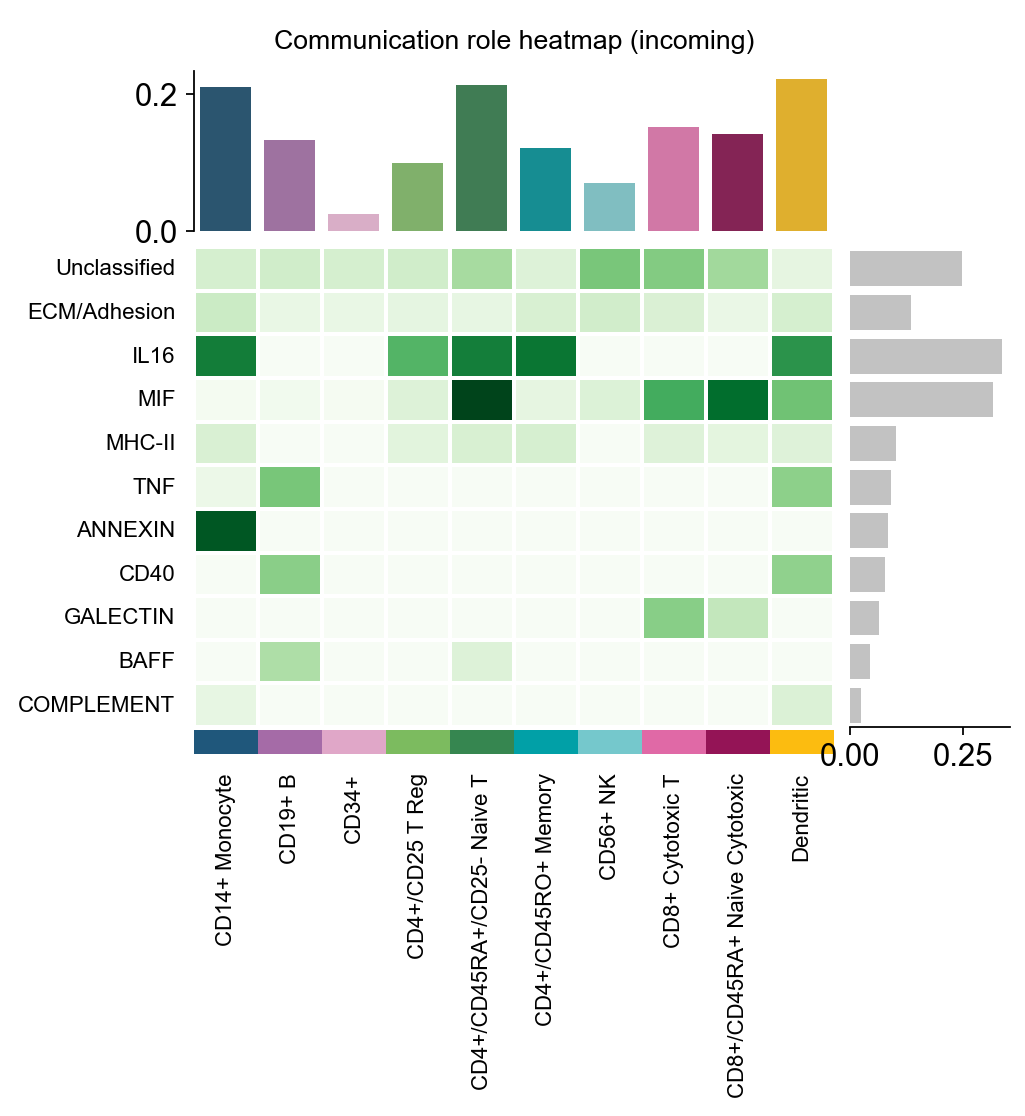
fig, ax = ov.pl.ccc_heatmap(
adata,
plot_type='role_heatmap',
pattern='incoming',
cmap='Greens',
figsize=(4, 3),
show=False,
)
🔬 Calculating cell communication strength for 11 pathways...
- Aggregation method: mean
- Minimum expression threshold: 0.1
✅ Completed pathway communication strength calculation for 11 pathways
📊 Pathway significance analysis results:
- Total pathways: 11
- Significant pathways: 11
- Strength threshold: 0.5
- p-value threshold: 0.05
🏆 Top 10 pathways by total strength:
----------------------------------------------------------------------------------------------------
Pathway Total Max Mean L-R Active Sig Rate Status
----------------------------------------------------------------------------------------------------
Unclassified 58.62 0.99 0.72 5 81 33 0.41 ***
ECM/Adhesion 35.78 1.00 0.76 6 47 25 0.53 ***
IL16 33.61 0.98 0.86 1 39 10 0.26 ***
MIF 31.59 0.98 0.64 1 49 5 0.10 ***
MHC-II 22.20 1.00 0.69 18 32 16 0.50 ***
TNF 20.53 1.00 0.89 3 23 14 0.61 ***
ANNEXIN 8.82 1.00 0.88 2 10 6 0.60 ***
CD40 7.79 1.00 0.97 1 8 7 0.88 ***
GALECTIN 6.39 1.00 0.58 1 11 4 0.36 ***
BAFF 5.83 1.00 0.97 2 6 5 0.83 ***
📊 Heatmap statistics:
- Number of pathways: 11
- Number of cell types: 10
- Signal strength range: 0.000 - 0.900
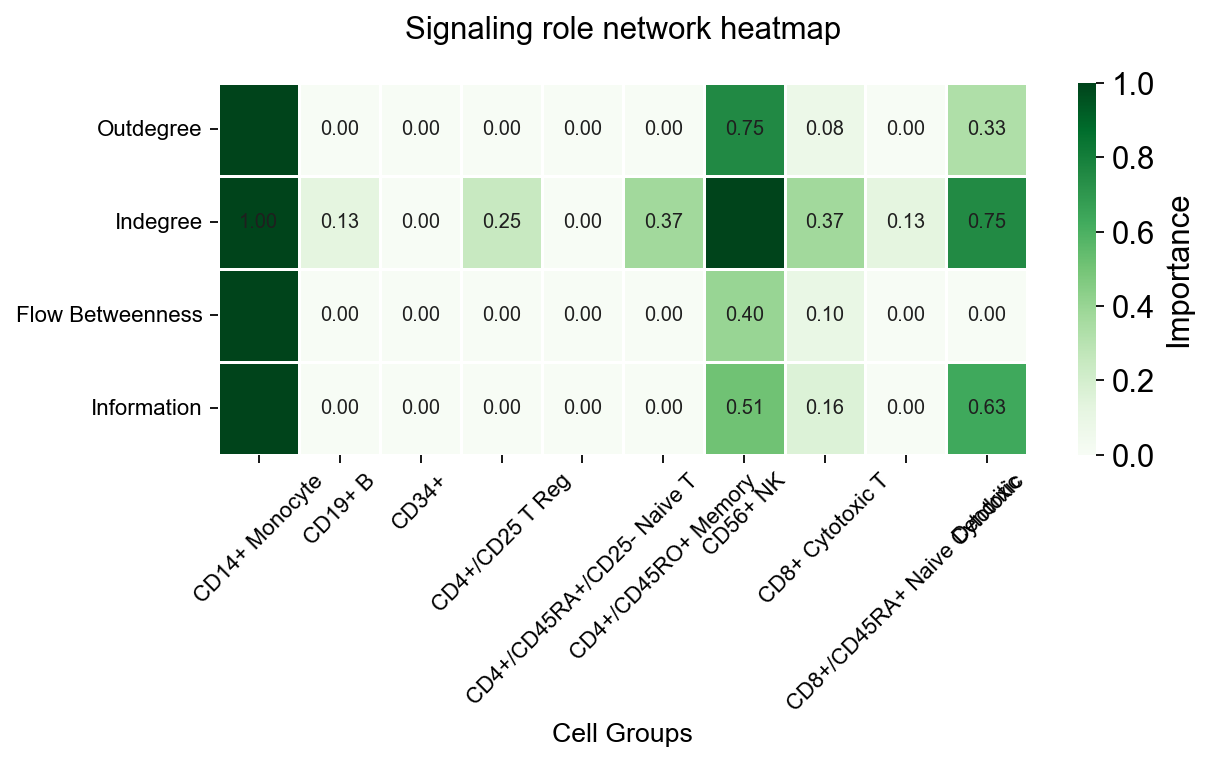
fig, ax = ov.pl.ccc_heatmap(
adata,
plot_type='role_network',
signaling=['ECM/Adhesion'],
cmap='Greens',
figsize=(8, 5),
show=False,
)
✅ Network centrality calculation completed (CellChat-style Importance values)
- Signaling pathways used: ['ECM/Adhesion']
- Weight mode: Weighted
- Calculated metrics: outdegree, indegree, flow_betweenness, information, overall
- All centrality scores normalized to 0-1 range (Importance values)
📊 Signaling role analysis results (Importance values 0-1):
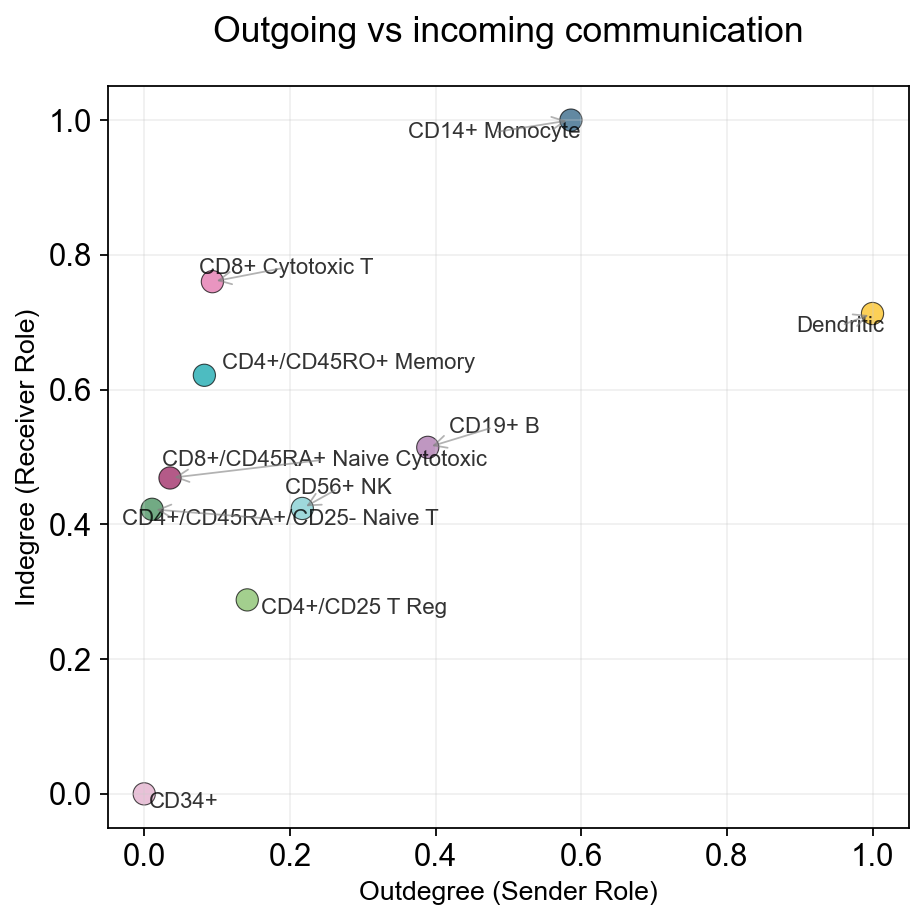
- Dominant Sender: CD14+ Monocyte (Importance: 1.000)
- Dominant Receiver: CD56+ NK (Importance: 1.000)
- Mediator: CD14+ Monocyte (Importance: 1.000)
- Influencer: CD14+ Monocyte (Importance: 1.000)
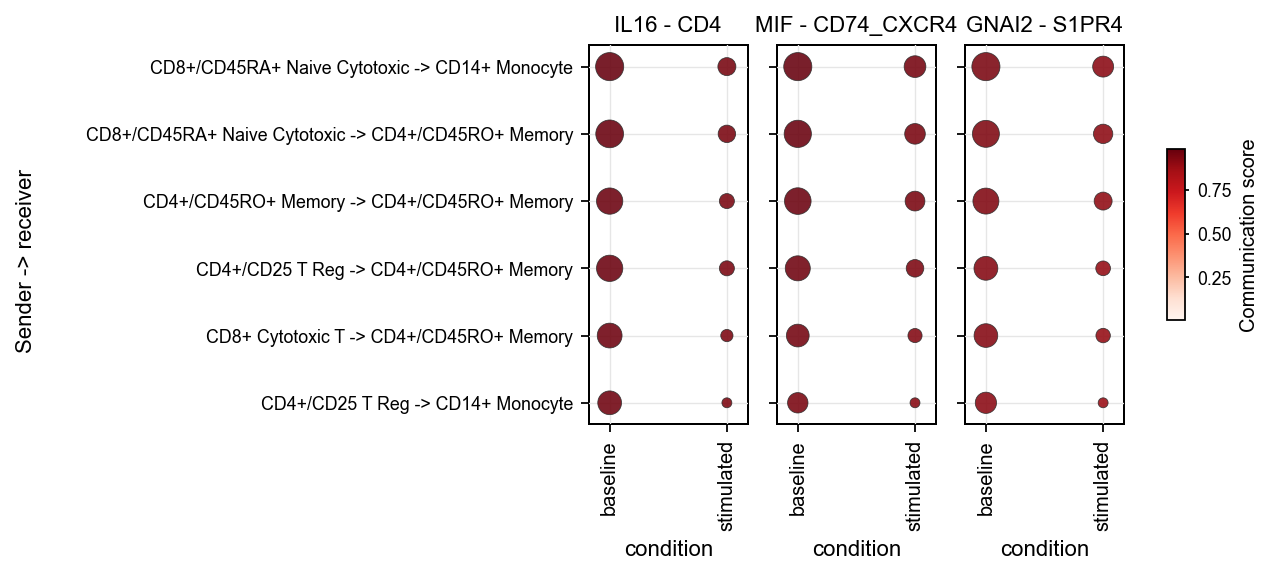
1.7 Sample/context dot plot#
sample_dot requires a context/sample column in the LIANA result table.
We first demonstrate the API with a synthetic tutorial-style condition: duplicate the same liana_res twice and manually add baseline / stimulated.
This step is only for showing the plotting API. It is not a real condition-comparison analysis.
adata_by_context = adata.copy()
frames = []
for condition, delta in [('baseline', 0.0), ('stimulated', 0.03)]:
frame = adata.uns['liana_res'].copy()
frame['condition'] = condition
frame['specificity_rank'] = np.clip(frame['specificity_rank'] + delta, 0.0, 1.0)
frames.append(frame)
adata_by_context.uns['liana_res'] = pd.concat(frames, ignore_index=True)
fig, ax = ov.pl.ccc_heatmap(
adata_by_context,
plot_type='sample_dot',
display_by='interaction',
score_key='specificity_rank',
pvalue_key='specificity_rank',
sample_key='condition',
top_n=3,
top_n_pairs=6,
pvalue_threshold=0.05,
figsize=(9, 3),
show=False,
)
If you have a real condition column, the correct workflow is to run LIANA separately per condition and then concatenate the result tables before calling sample_dot. A minimal template is shown below.
# condition_key = 'condition'
# result_frames = []
# for condition in sorted(adata.obs[condition_key].dropna().astype(str).unique()):
# ad_sub = adata[adata.obs[condition_key].astype(str) == condition].copy()
# ov.single.run_liana(
# ad_sub,
# groupby='bulk_labels',
# method='rank_aggregate',
# key_added='liana_res',
# inplace=True,
# )
# frame = ad_sub.uns['liana_res'].copy()
# frame[condition_key] = condition
# result_frames.append(frame)
# adata_real_context = adata.copy()
# adata_real_context.uns['liana_res'] = pd.concat(result_frames, ignore_index=True)
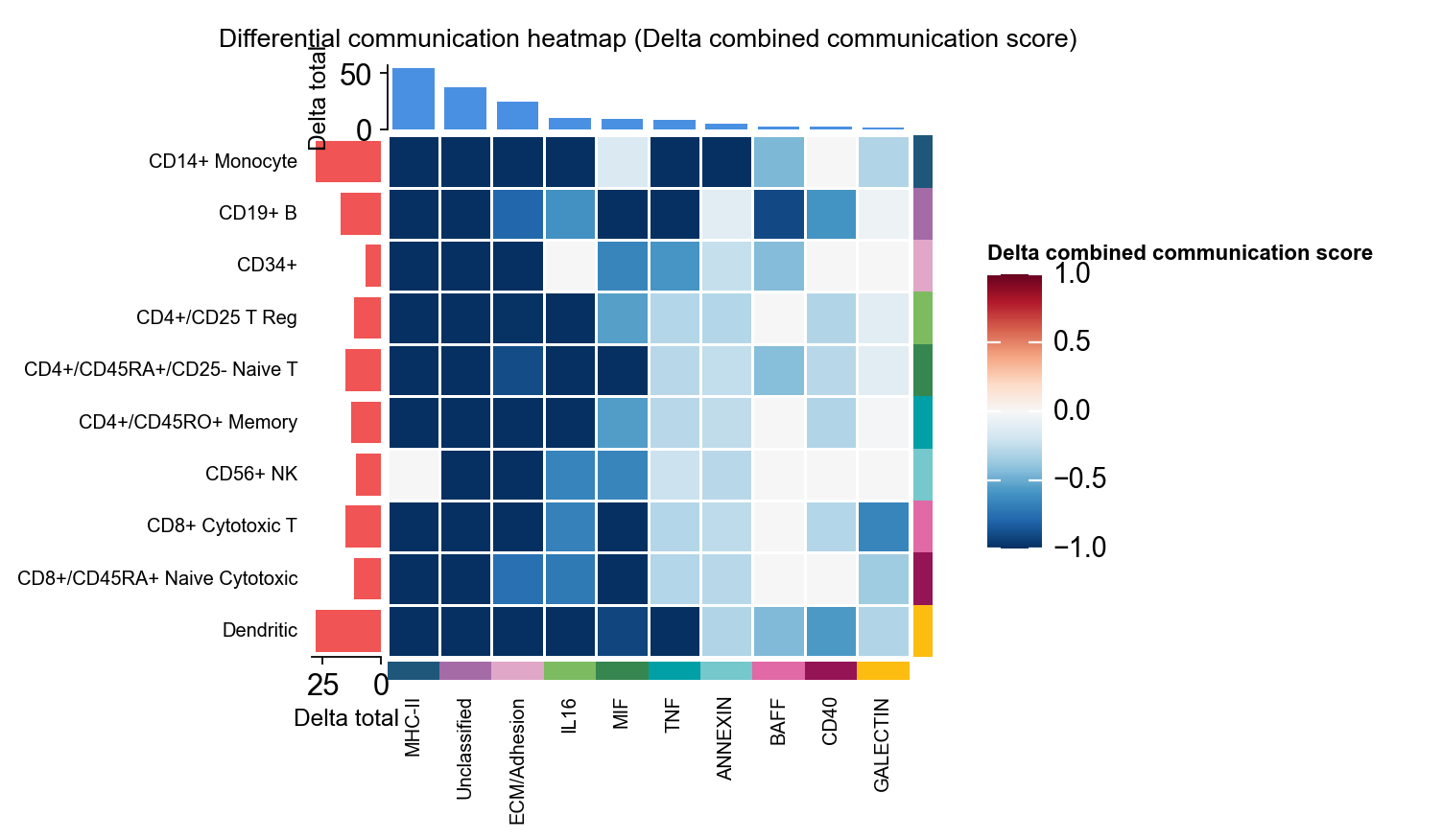
1.8 Differential heatmap#
To demonstrate the differential heatmap, we construct a synthetic comparison object directly from comm_adata.
fig, ax = ov.pl.ccc_heatmap(
adata,
comparison_adata=comparison_comm,
plot_type='diff_heatmap',
top_n=10,
figsize=(5,5),
show=False,
show_col_names=True,
show_row_names=True,
)
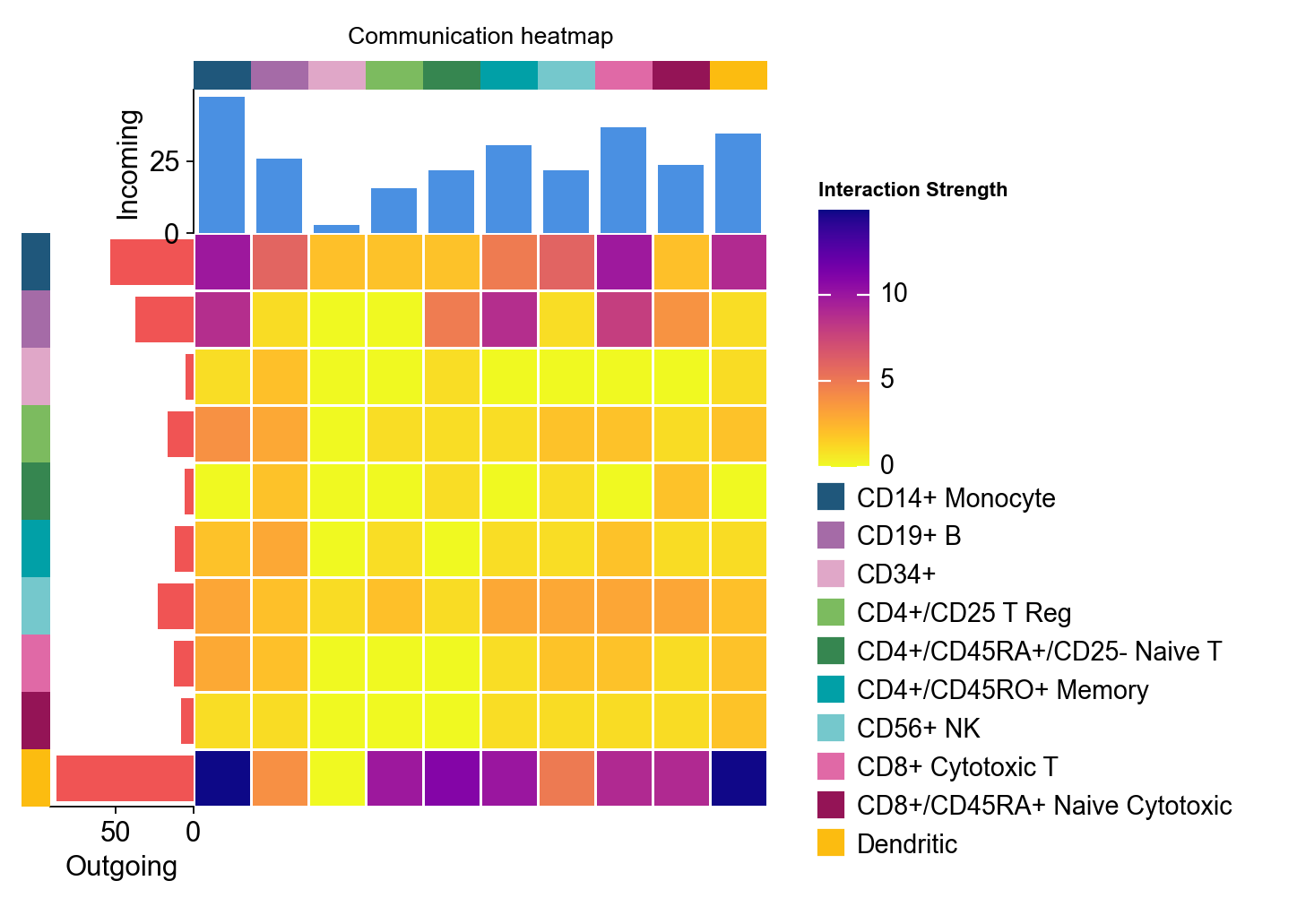
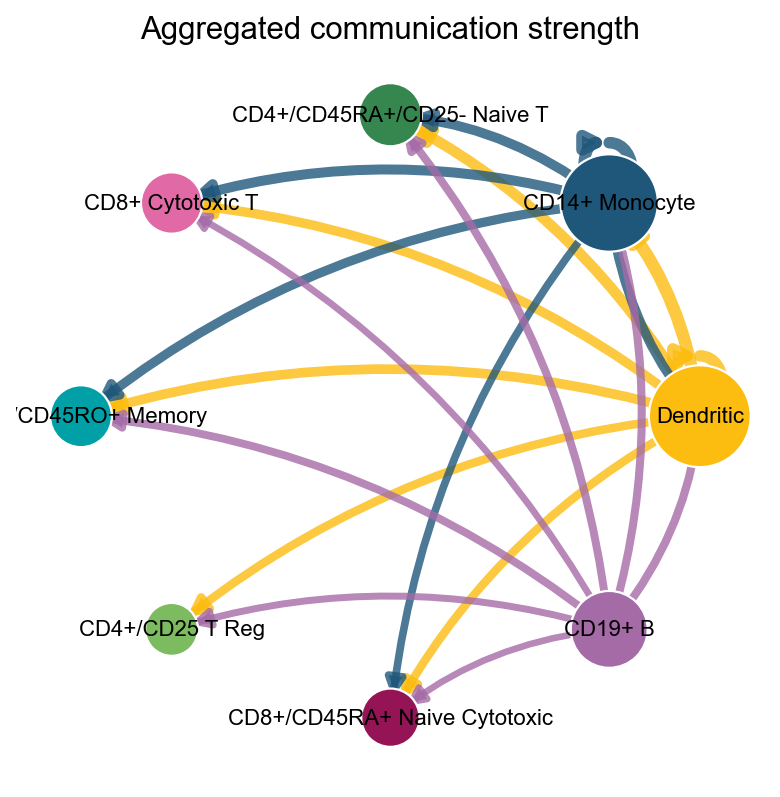
2. ov.pl.ccc_network_plot#
Heatmaps are good for matrix structure. Network plots are better for questions such as:
which cell groups act as dominant senders or receivers
what the overall direction of one pathway looks like
where a specific ligand or receptor sits in the communication graph
fig, ax = ov.pl.ccc_network_plot(
adata,
plot_type='circle',
palette=color_dict,
title='Aggregated communication strength',
figsize=(6, 6),
show=False,
)
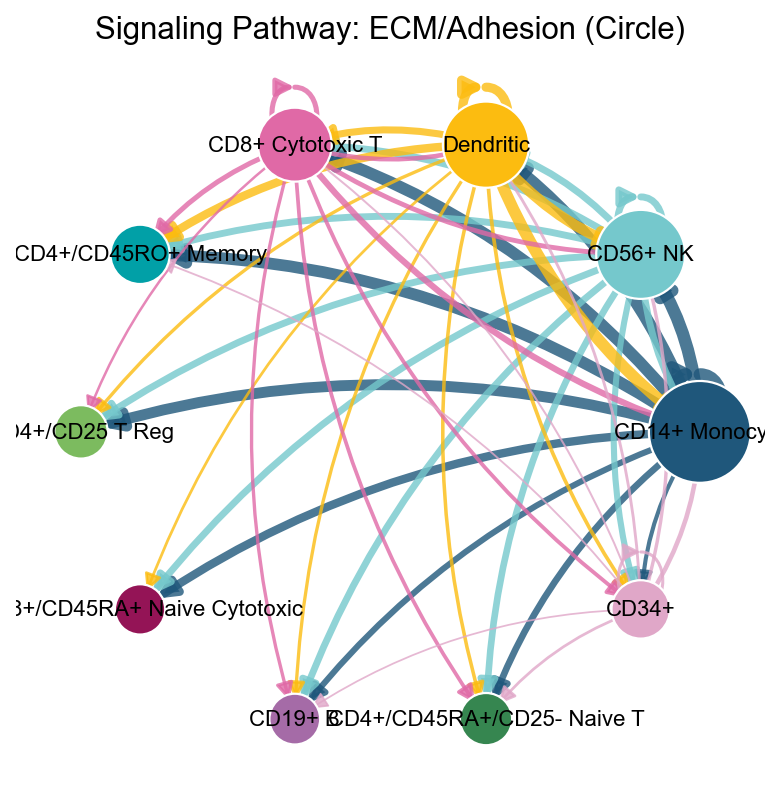
fig, ax = ov.pl.ccc_network_plot(
adata,
plot_type='pathway',
signaling=['ECM/Adhesion'],
palette=color_dict,
top_n=50,
figsize=(6, 6),
show=False,
)
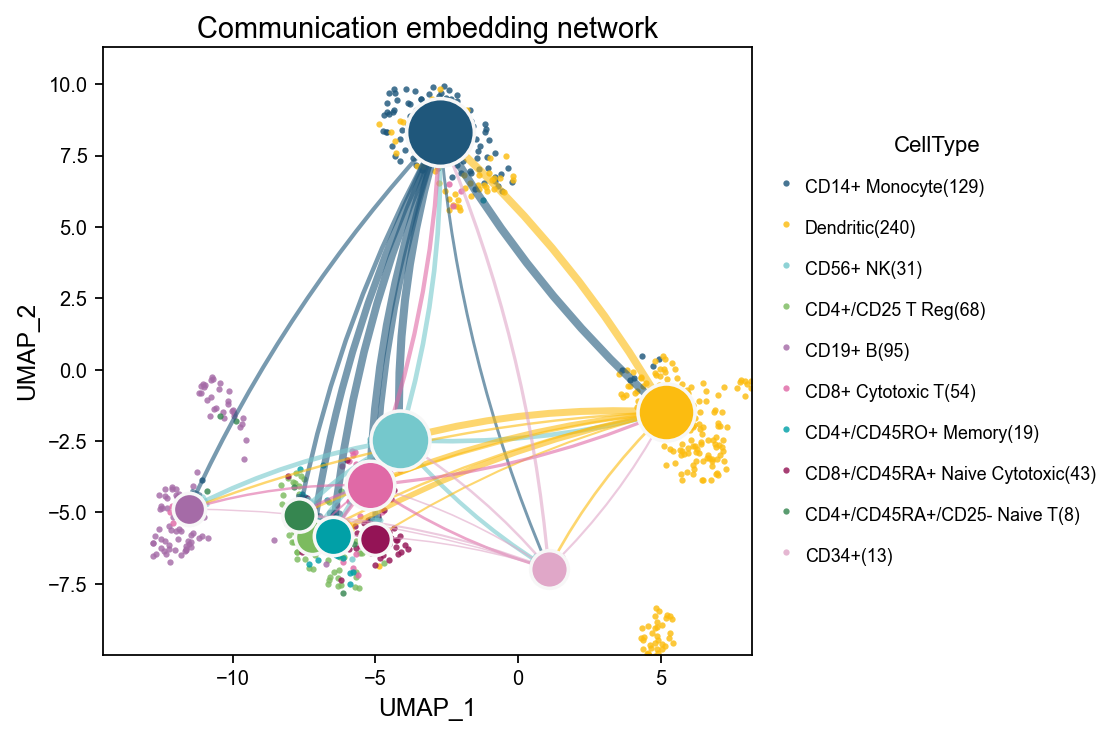
embedding_network overlays the communication network on a low-dimensional embedding. Background points show the distribution of cells in transcriptomic space, larger nodes represent aggregated cell-type nodes, and edges show the direction and strength of communication. This is useful when you want to inspect whether stronger communication tends to occur between nearby or functionally related cell groups in embedding space.
fig, ax = ov.pl.ccc_network_plot(
adata,
plot_type='embedding_network',
signaling=['ECM/Adhesion'],
node_positions=node_positions,
embedding_points=embedding_points,
palette=color_dict,
top_n=50,
figsize=(6, 5),
show=False,
)
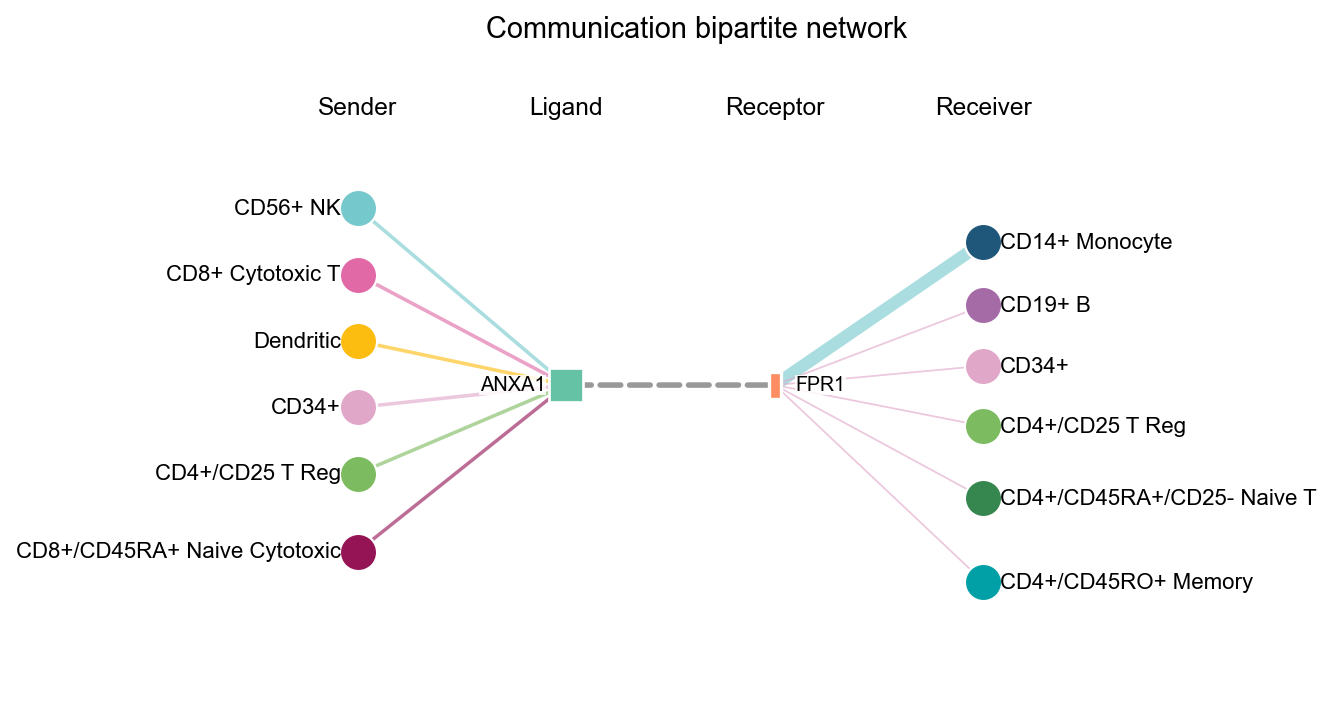
fig, ax = ov.pl.ccc_network_plot(
adata,
plot_type='bipartite',
ligand=focus_ligand,
palette=color_dict,
top_n=6,
figsize=(6, 5),
show=False,
)
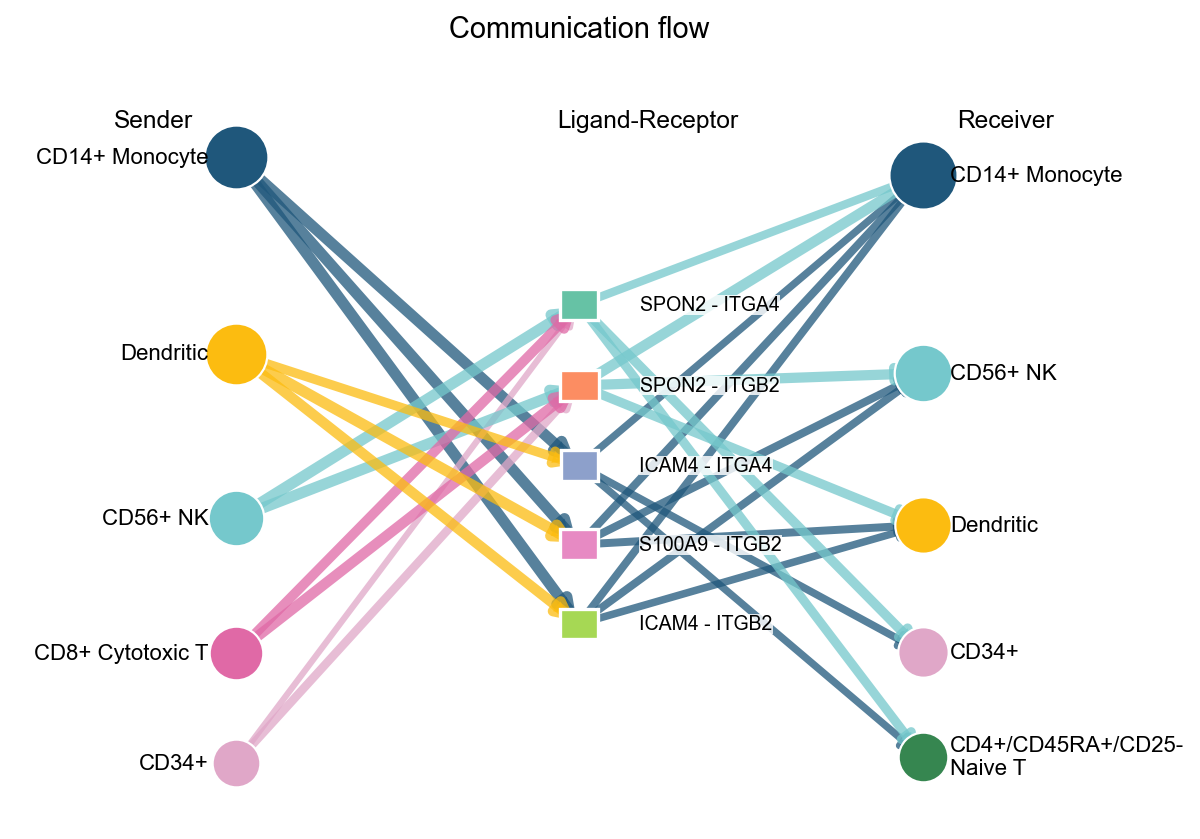
fig, ax = ov.pl.ccc_network_plot(
adata,
plot_type='arrow',
display_by='interaction',
signaling=['ECM/Adhesion'],
palette=color_dict,
top_n=5,
figsize=(8, 6),
show=False,
)
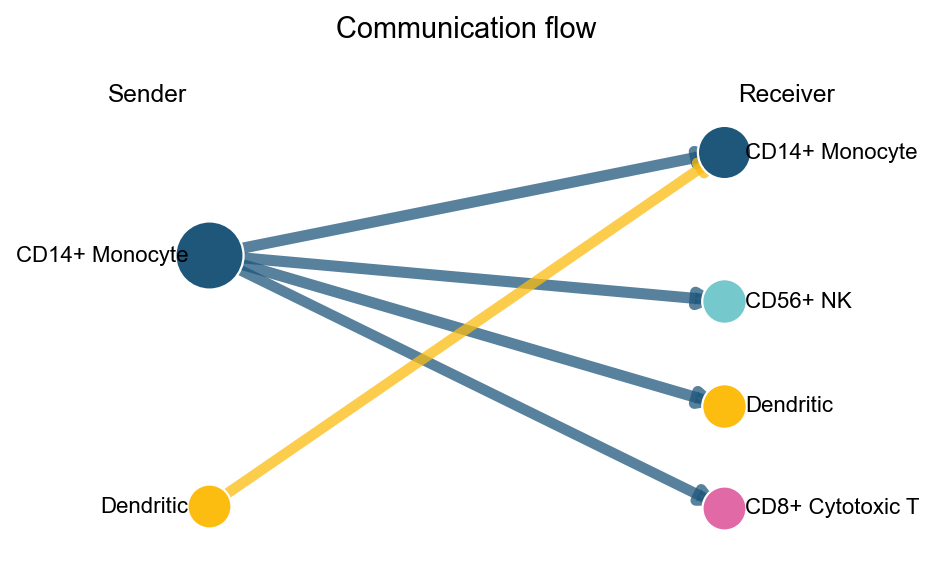
arrow and sigmoid also support display_by='aggregation'. In that mode, the plot only keeps sender-receiver pathway relations, and top_n controls how many relations are shown.
fig, ax = ov.pl.ccc_network_plot(
adata,
plot_type='arrow',
display_by='aggregation',
signaling=['ECM/Adhesion'],
palette=color_dict,
top_n=5,
figsize=(6, 4),
show=False,
)
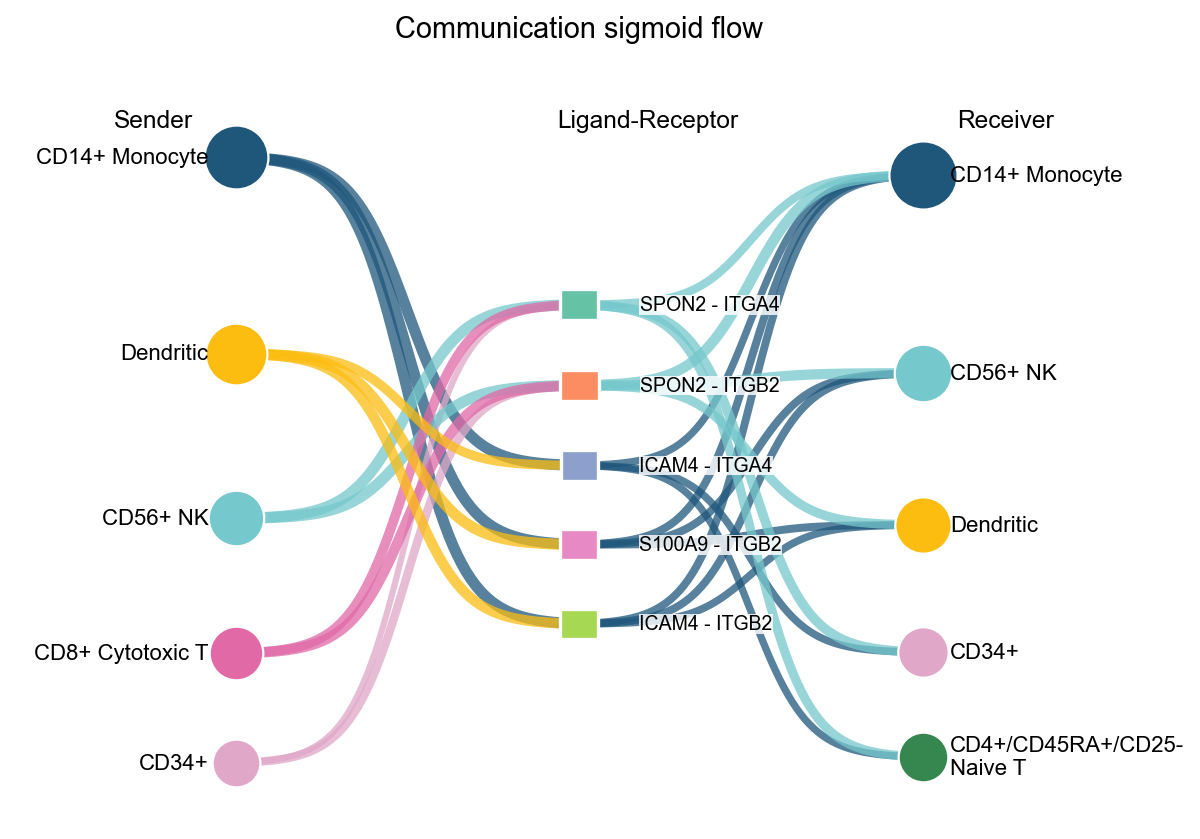
fig, ax = ov.pl.ccc_network_plot(
adata,
plot_type='sigmoid',
display_by='interaction',
signaling=['ECM/Adhesion'],
palette=color_dict,
top_n=5,
figsize=(8, 6),
show=False,
)
fig, ax = ov.pl.ccc_network_plot(
adata,
plot_type='individual_outgoing',
figsize=(12, 15),
ncols=3,
show=False,
)
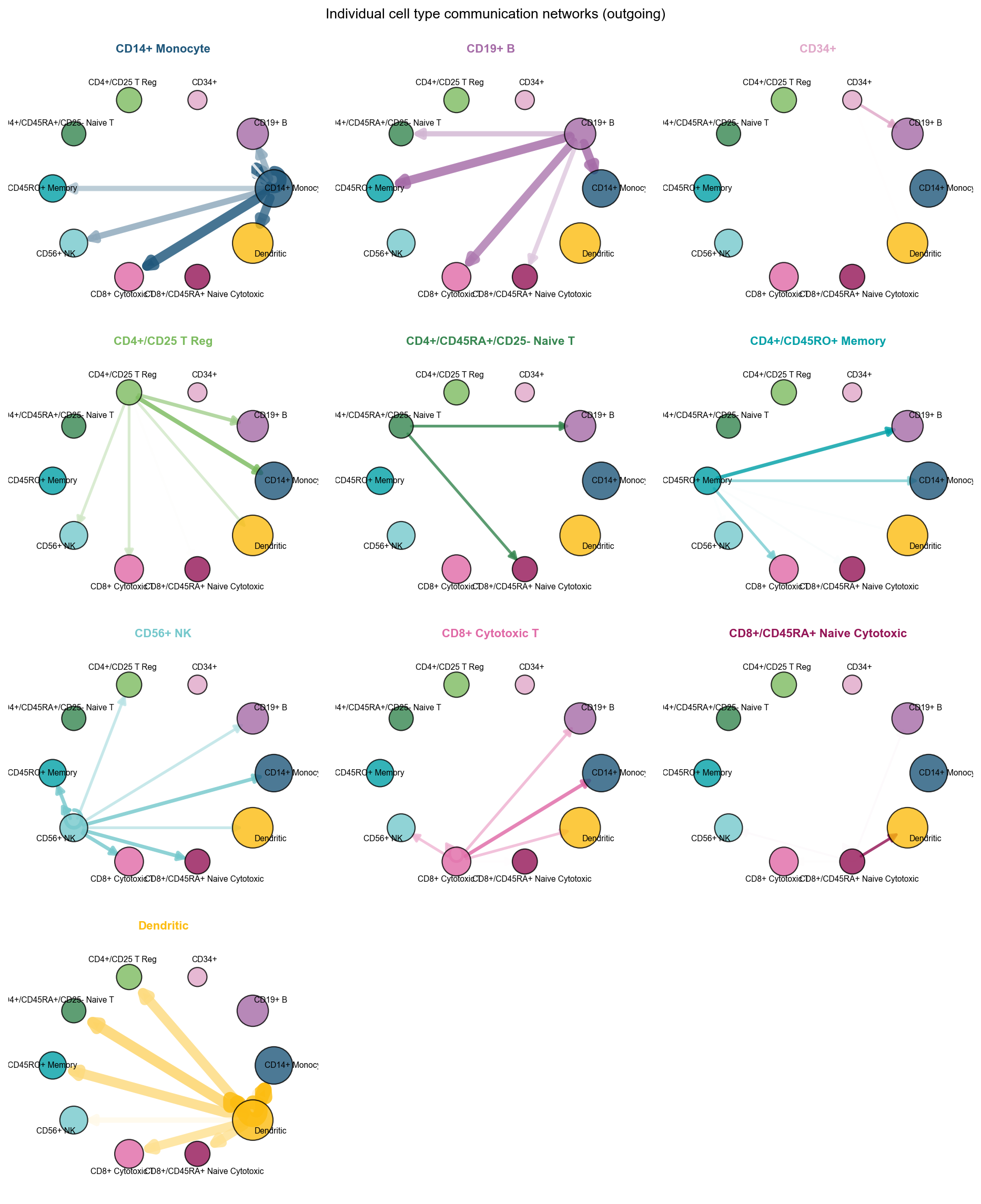
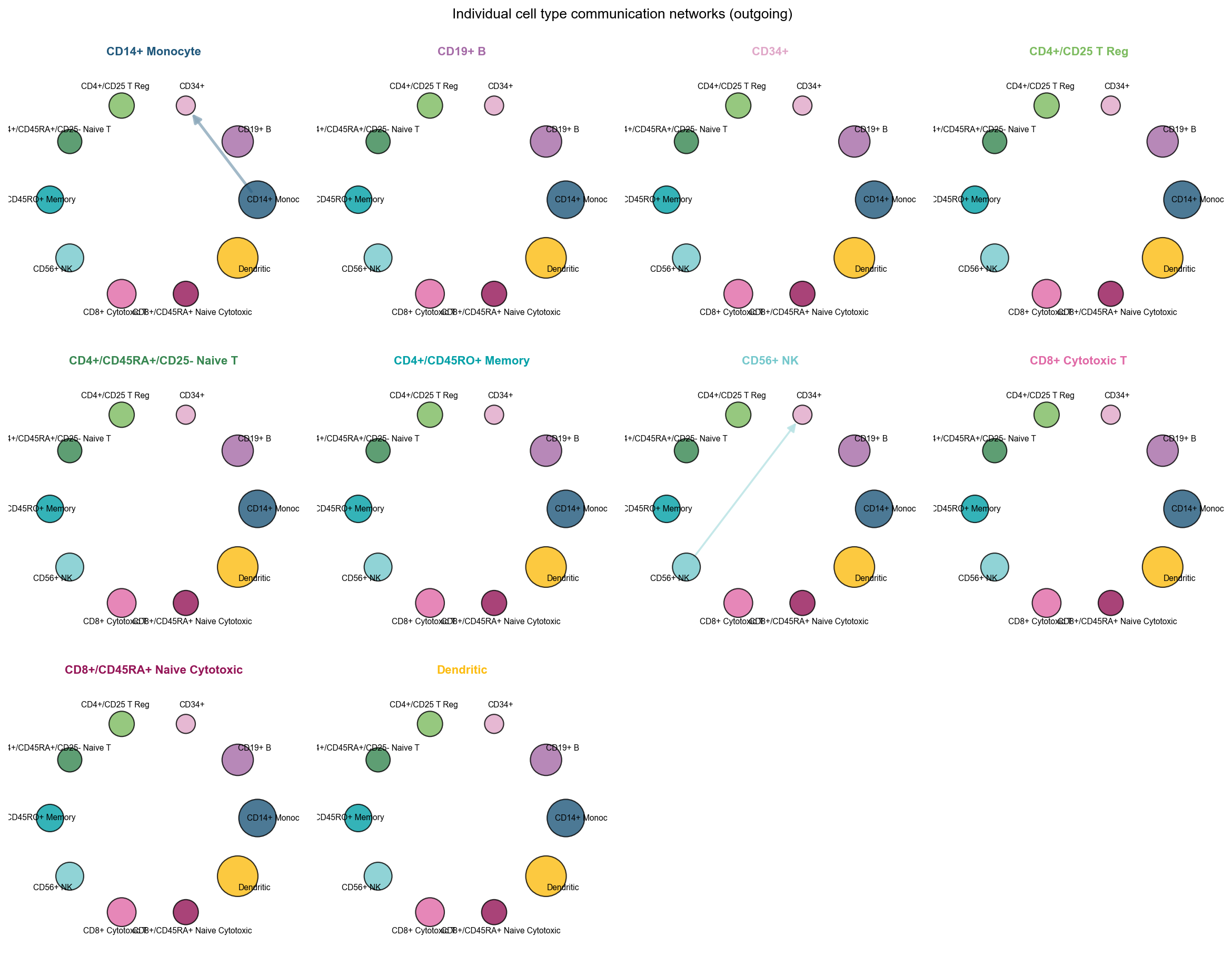
fig, ax = ov.pl.ccc_network_plot(
adata,
plot_type='individual_outgoing',
receiver_use='CD34+',
figsize=(15, 12),
show=False,
)
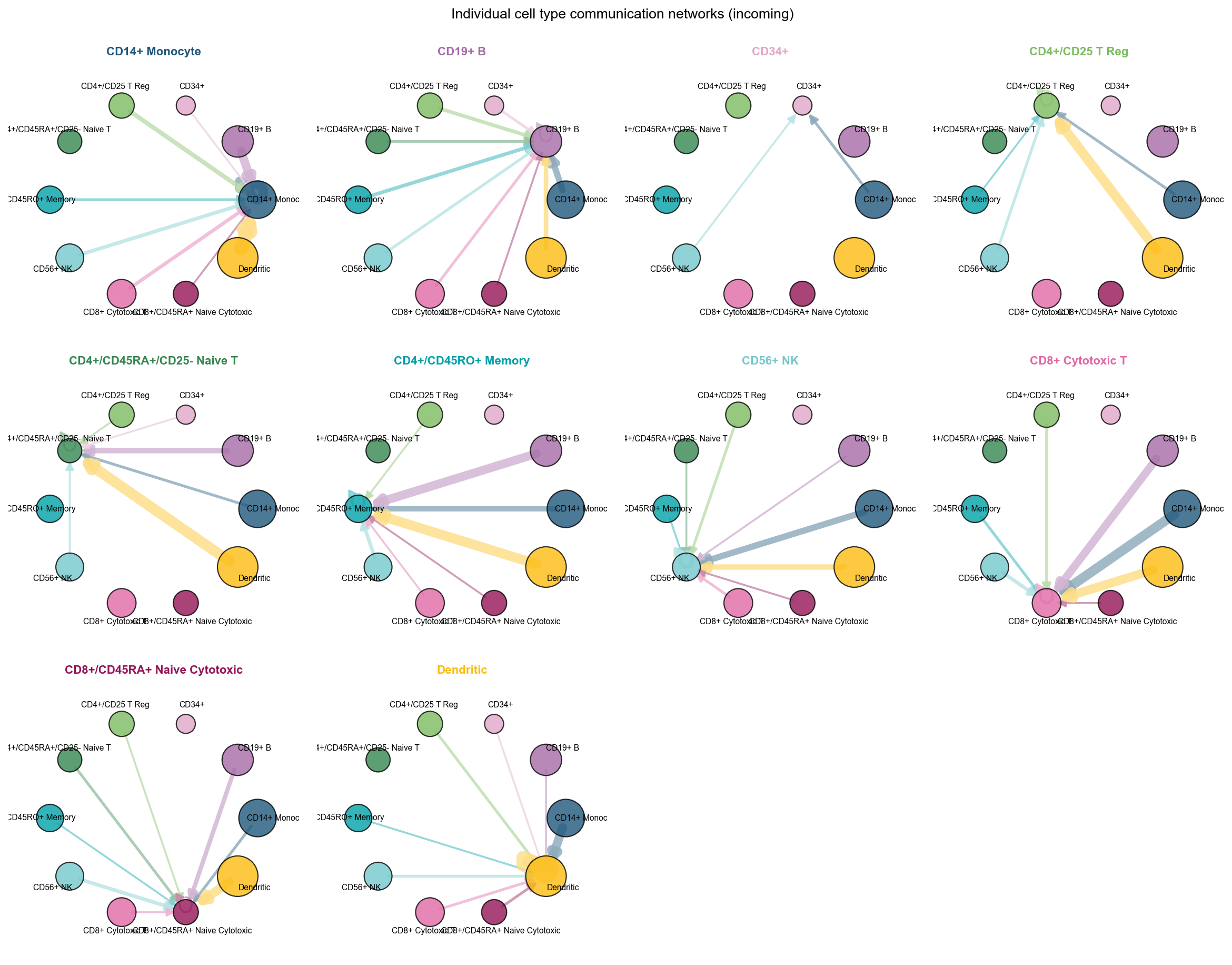
fig, ax = ov.pl.ccc_network_plot(
adata,
plot_type='individual_incoming',
figsize=(15, 12),
show=False,
)
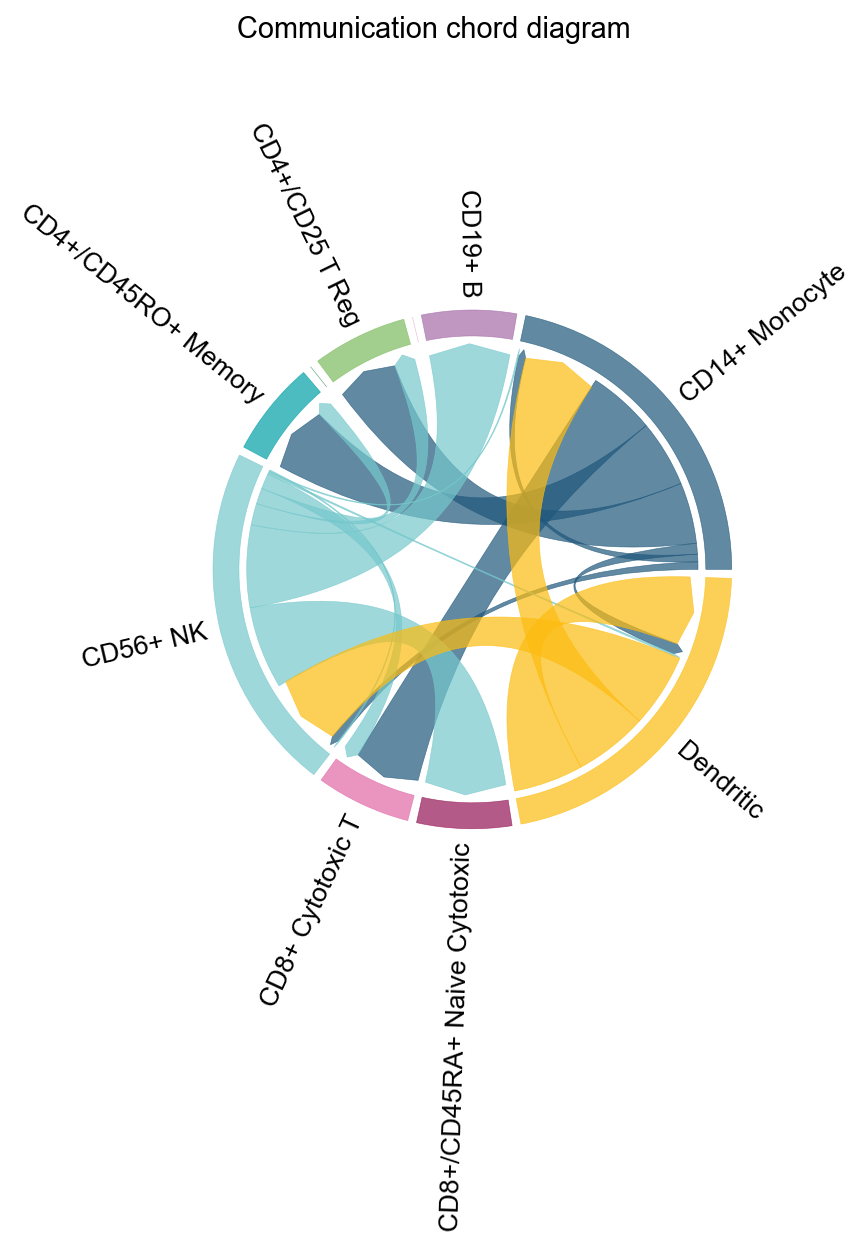
fig, ax = ov.pl.ccc_network_plot(
adata,
plot_type='chord',
signaling=['ECM/Adhesion'],
normalize_to_sender=True,
figsize=(8, 8),
show=False,
)
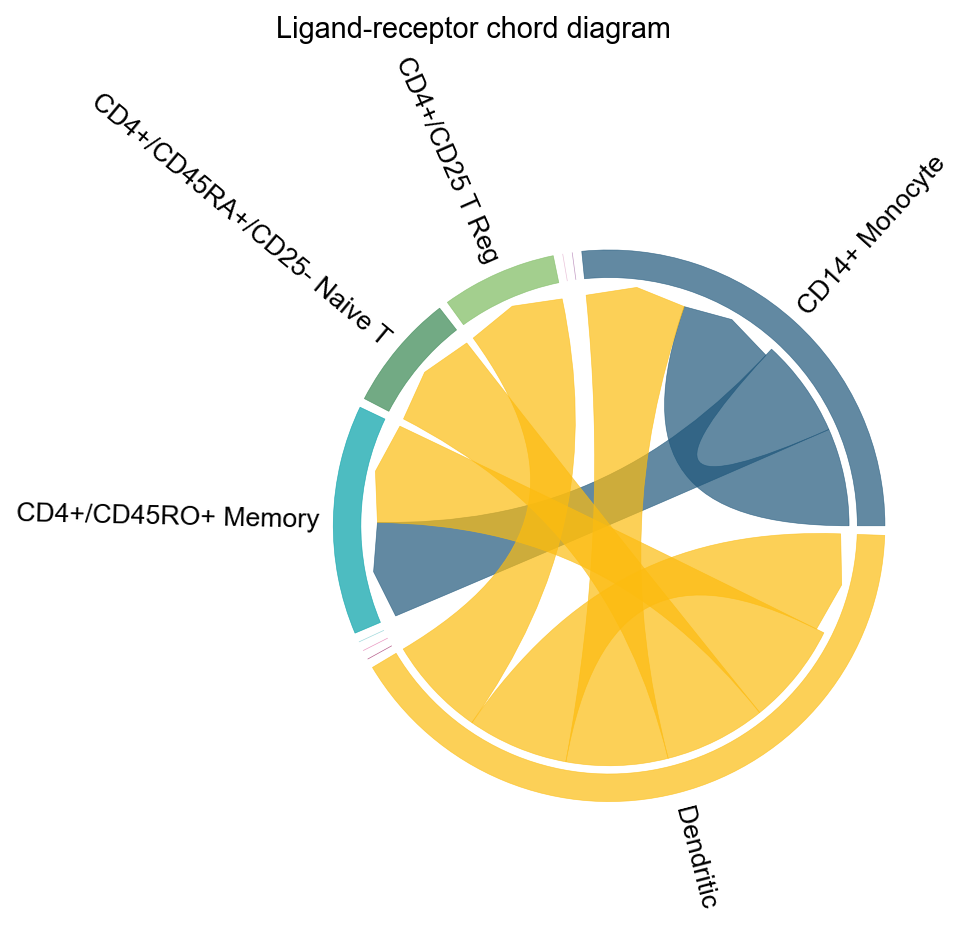
fig, ax = ov.pl.ccc_network_plot(
adata,
plot_type='lr_chord',
pair_lr_use=focus_pair_lr,
palette=color_dict,
figsize=(6, 6),
show=False,
)
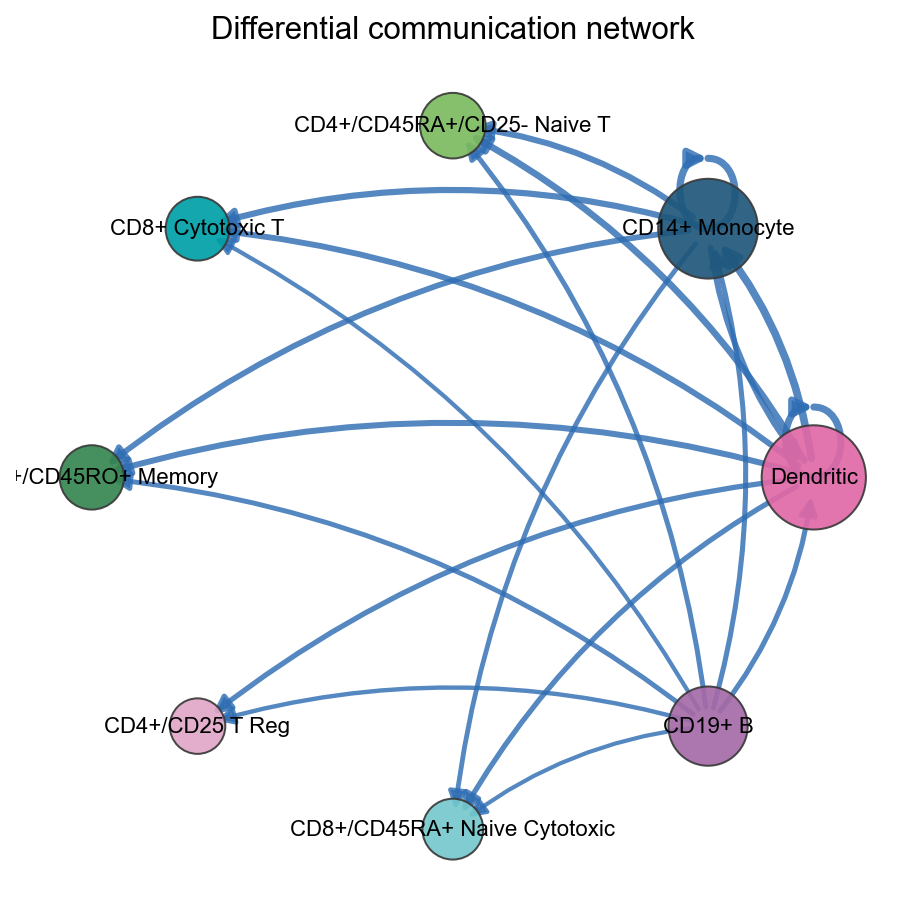
fig, ax = ov.pl.ccc_network_plot(
adata,
comparison_adata=comparison_comm,
plot_type='diff_network',
top_n=20,
show=False,
)
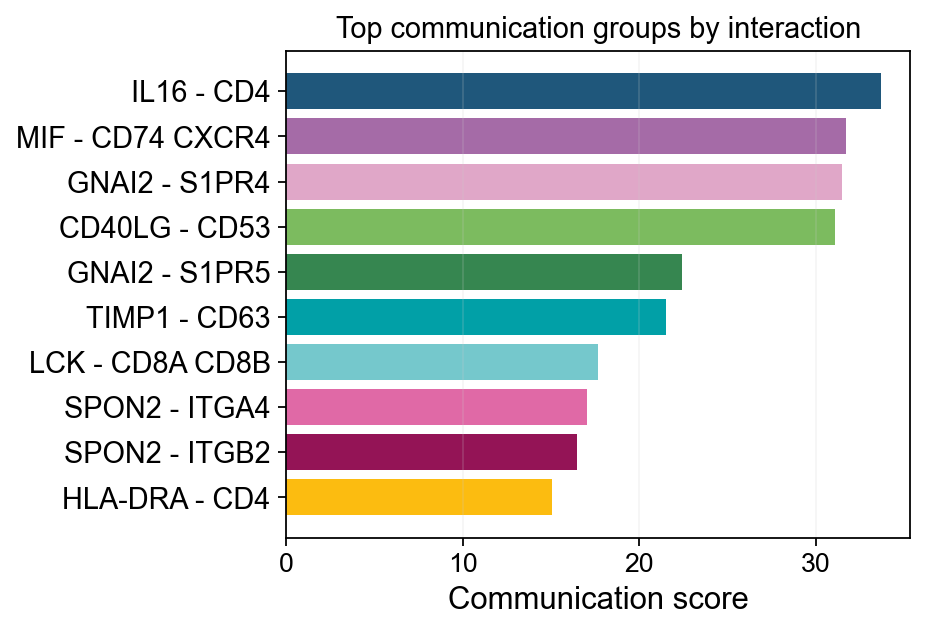
3. ov.pl.ccc_stat_plot#
Statistical summary plots are used for ranking, compression, and explanation:
bar: the most compact interaction/pathway rankingscatter: overview of communication distributionssankey: sender/receiver/interaction flow structurepathway_summary: pathway-level total strength plus significant pair countslr_contribution: which ligand-receptor pairs explain one pathway
fig, ax = ov.pl.ccc_stat_plot(
adata,
plot_type='bar',
display_by='interaction',
score_key='specificity_rank',
pvalue_key='specificity_rank',
group_by='interaction',
top_n=10,
figsize=(5, 4),
show=False,
)
fig, ax = ov.pl.ccc_stat_plot(
adata,
plot_type='scatter',
figsize=(6, 6),
show=False,
)
✅ Network centrality calculation completed (CellChat-style Importance values)
- Signaling pathways used: All pathways
- Weight mode: Weighted
- Calculated metrics: outdegree, indegree, flow_betweenness, information, overall
- All centrality scores normalized to 0-1 range (Importance values)
fig, ax = ov.pl.ccc_stat_plot(
adata,
plot_type='sankey',
display_by='interaction',
signaling=['ECM/Adhesion'],
figsize=(8, 6),
show=False,
)
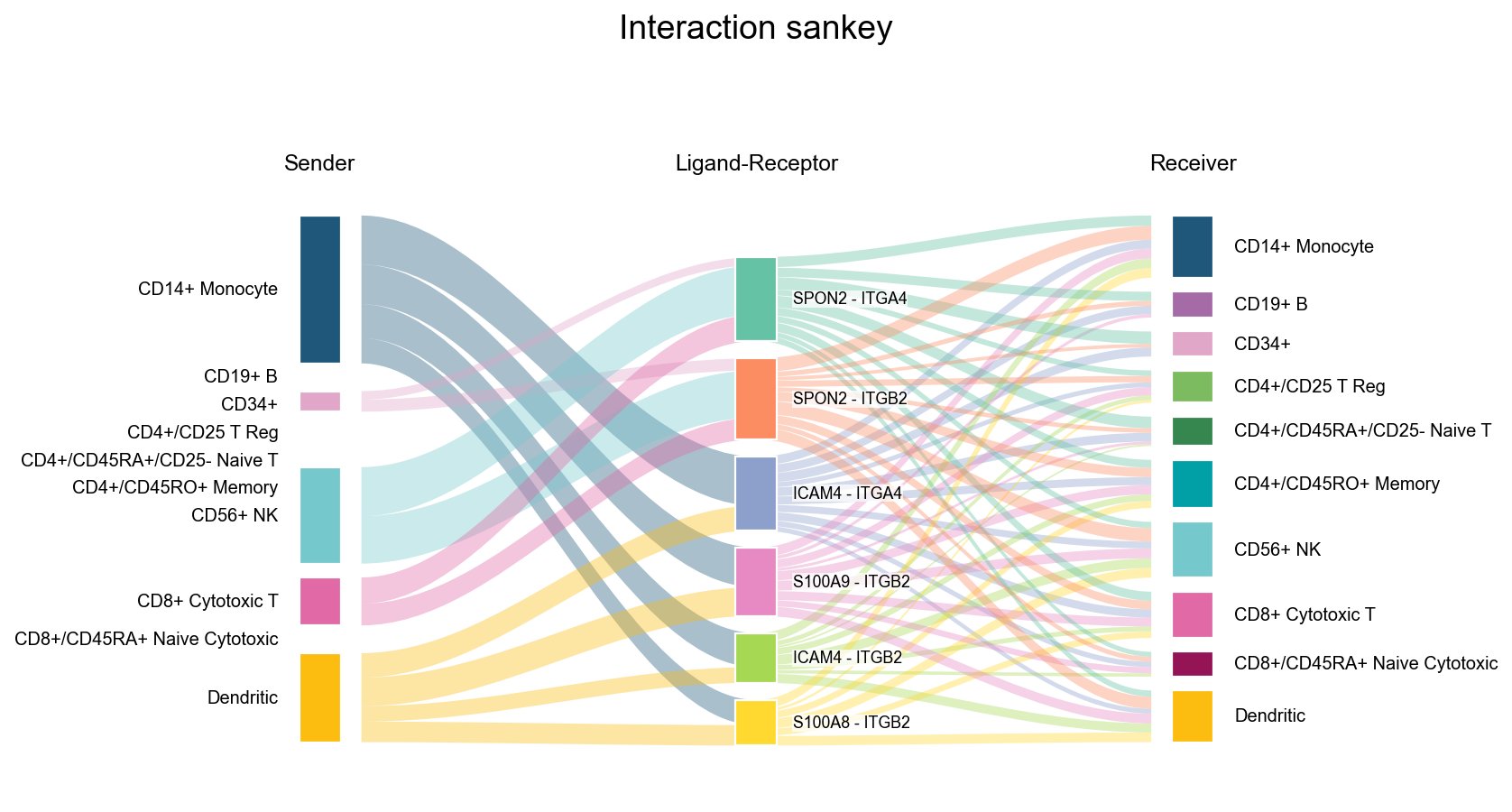
fig, ax = ov.pl.ccc_stat_plot(
adata,
plot_type='sankey',
display_by='aggregation',
figsize=(6, 4),
show=False,
)
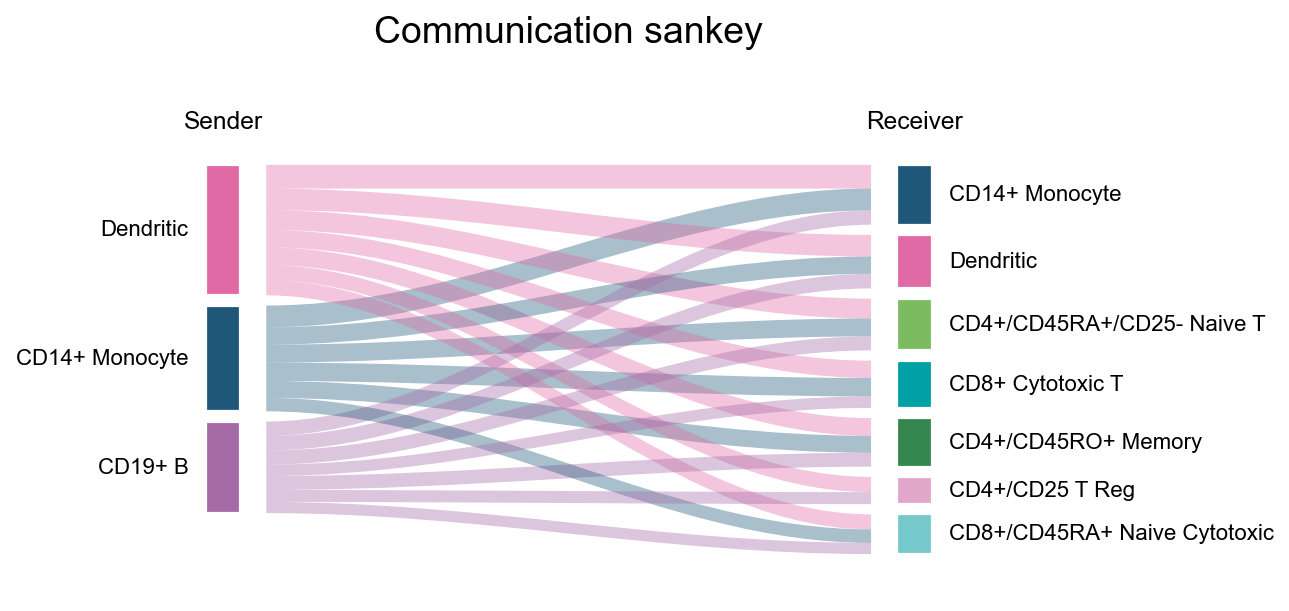
fig, ax = ov.pl.ccc_stat_plot(
adata,
plot_type='pathway_summary',
score_key='specificity_rank',
pvalue_key='specificity_rank',
classification_reference='cellchat',
classification_fallback='family',
top_n=10,
pvalue_threshold=0.05,
min_expression=0.0,
strength_threshold=0.0,
min_significant_pairs=1,
figsize=(4, 4),
verbose=True,
show=False,
)
🔬 Calculating cell communication strength for 11 pathways...
- Aggregation method: mean
- Minimum expression threshold: 0.0
✅ Completed pathway communication strength calculation for 11 pathways
📊 Pathway significance analysis results:
- Total pathways: 11
- Significant pathways: 11
- Strength threshold: 0.0
- p-value threshold: 0.05
🏆 Top 10 pathways by total strength:
----------------------------------------------------------------------------------------------------
Pathway Total Max Mean L-R Active Sig Rate Status
----------------------------------------------------------------------------------------------------
IL16 33.69 0.98 0.84 1 40 10 0.25 ***
MIF 31.72 0.98 0.62 1 51 5 0.10 ***
Unclassified 24.84 0.67 0.30 5 82 33 0.40 ***
ECM/Adhesion 13.60 0.67 0.29 6 47 25 0.53 ***
MHC-II 10.13 0.55 0.31 18 33 16 0.48 ***
TNF 9.02 0.91 0.38 3 24 14 0.58 ***
ANNEXIN 8.44 1.00 0.84 2 10 6 0.60 ***
CD40 7.79 1.00 0.97 1 8 7 0.88 ***
GALECTIN 6.39 1.00 0.58 1 11 4 0.36 ***
BAFF 4.42 1.00 0.74 2 6 5 0.83 ***
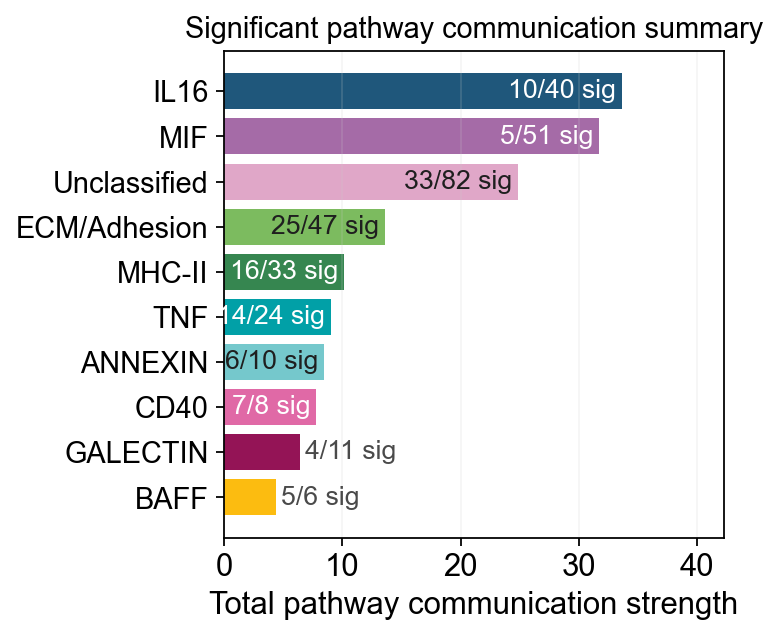
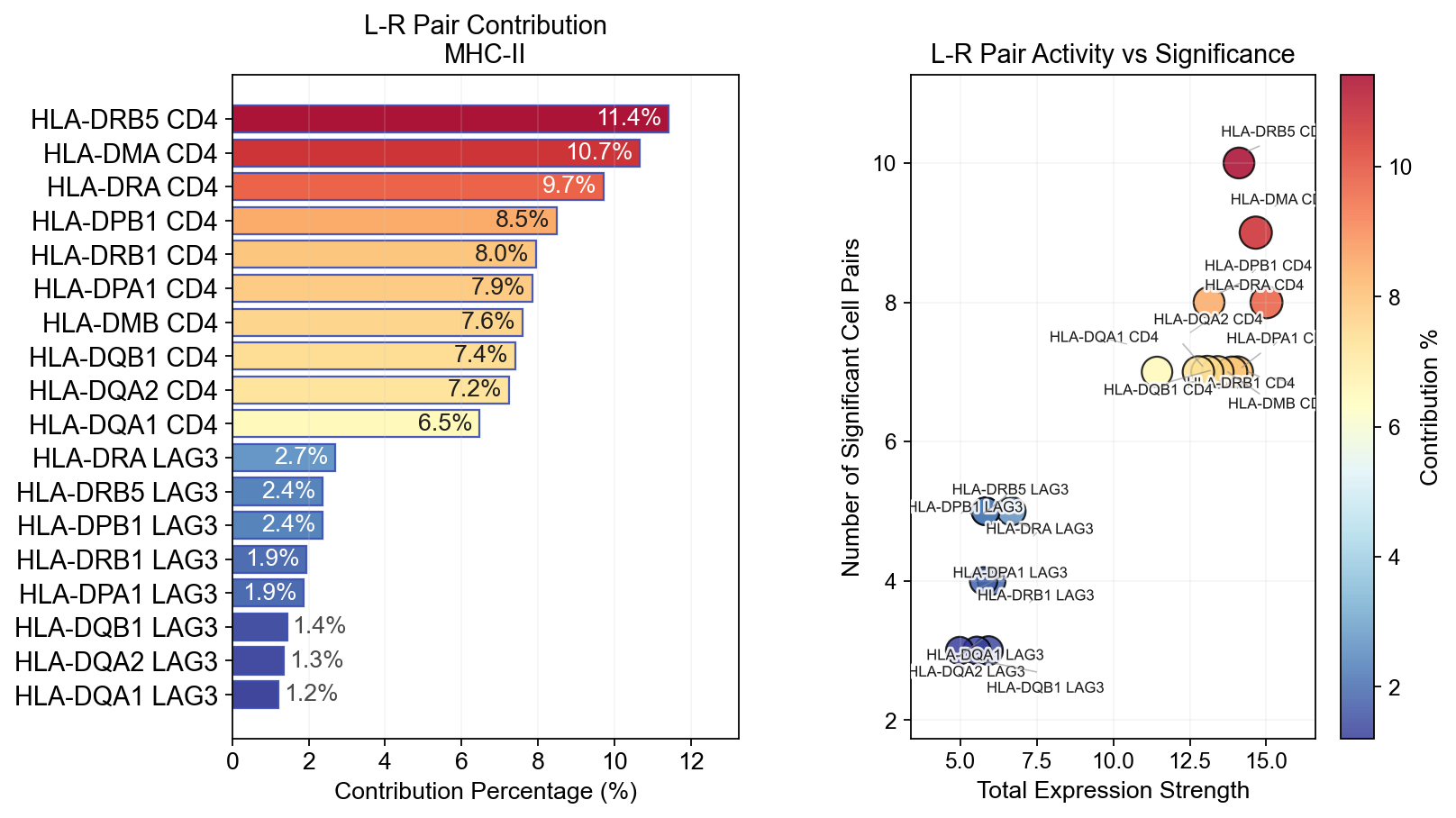
fig, ax = ov.pl.ccc_stat_plot(
adata,
plot_type='lr_contribution',
signaling=['MHC-II'],
figsize=(10, 6),
show=False,
)