Single-cell copy-number variation with CopyKAT#
This tutorial walks through ov.single.CNV(method='copykat') — a thin
omicverse wrapper around the pure-Python
py-CopyKAT re-implementation of
the R CopyKAT method
(Gao et al., Nature Biotechnology 2021).
CopyKAT is unsupervised: it classifies each cell as aneuploid
(tumour) or diploid (normal) without needing a reference cell-type
annotation. The wrapper writes results to a unified schema shared with
inferCNV so the plotting helpers (ov.pl.cnv_heatmap, ov.pl.cnv_summary,
ov.pl.cnv_umap) work with either backend.
Part.1 The math behind CopyKAT#
CopyKAT runs an 11-step pipeline. The core ideas:
1. Stabilise raw counts. The variance-stabilising transform $\(\;\;y = \log(\sqrt{x} + \sqrt{x+1})\)$ maps Poisson-distributed counts to a near-Gaussian scale, after which the expression matrix is centred per cell.
2. Local denoising. A Kalman smoother sweeps along each chromosome to suppress per-gene noise while preserving step changes at copy-number breakpoints.
3. Baseline estimation. A Gaussian mixture (or the user-supplied
norm_cell_names) identifies the diploid baseline, which is subtracted
to give per-gene log-ratios.
4. Segmentation. A Poisson-gamma MCMC scheme detects breakpoints shared across cells, then aggregates genes into ~220 kb bins.
5. Classification. A KS-test on the aggregated CN profile flags each cell as \(\mathrm{aneuploid}\) (\(\Delta \mathrm{KS}\ge\tau\), default 0.1) or \(\mathrm{diploid}\). Aneuploid cells are then clustered by dynamic tree cut to recover subclones.
Reference: Gao et al., Nat Biotechnol (2021).
Part.2 Load data#
We use the Maynard et al. (2020) lung adenocarcinoma 3 000-cell scRNA-seq
benchmark. The file already carries gene-coordinate metadata in
adata.var (chromosome, start, end), which both CopyKAT and
inferCNV expect.
import omicverse as ov
ov.plot_set()
🔬 Starting plot initialization...
🧬 Detecting GPU devices…
✅ NVIDIA CUDA GPUs detected: 1
• [CUDA 0] NVIDIA H100 80GB HBM3
Memory: 79.1 GB | Compute: 9.0
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.1.3rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
import os
import scanpy as sc
import omicverse as ov
# Maynard et al. (2020) lung adenocarcinoma scRNA-seq — 3 000 cells, 55 556
# genes, gene coordinates already in adata.var. Bundled by infercnvpy.
DATA_URL = "https://github.com/icbi-lab/infercnvpy/releases/download/d0.1.0/maynard2020_3k.h5ad"
data_dir = "data/cnv"
os.makedirs(data_dir, exist_ok=True)
adata = sc.read(f"{data_dir}/maynard2020_3k.h5ad", backup_url=DATA_URL)
adata
AnnData object with n_obs × n_vars = 3000 × 55556
obs: 'age', 'sex', 'sample', 'patient', 'cell_type'
var: 'ensg', 'mito', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_counts', 'chromosome', 'start', 'end', 'gene_id', 'gene_name'
uns: 'cell_type_colors', 'neighbors', 'umap'
obsm: 'X_scVI', 'X_umap'
obsp: 'connectivities', 'distances'
# Quick look at cell-type composition. Epithelial cells are the
# candidate tumour population; immune/stromal cells (Macrophage, T cell,
# Fibroblast, ...) act as the diploid majority CopyKAT needs to anchor
# the baseline against.
adata.obs["cell_type"].value_counts().head(10)
cell_type
Epithelial cell 522
Macrophage 409
Fibroblast 308
T cell CD4 259
T cell CD8 246
Monocyte 227
Endothelial cell 148
Plasma cell 139
other 115
mDC 106
Name: count, dtype: int64
Part.3 Run CopyKAT#
ov.single.CNV(method='copykat') consumes raw counts directly. For the
3 000-cell input it finishes in ~1-2 minutes on CPU (pycopykat is a
Numba-accelerated port; no R / no GPU required).
After cnv.run():
slot |
content |
|---|---|
|
cells × ~12 k 220 kb bins of log-CN |
|
chromosome → starting-bin index |
|
|
|
per-cell mean abs(log-CN) |
cnv = ov.single.CNV(adata, method='copykat', genome='hg20')
cnv.run(verbose=False)
<omicverse.single._cnv.CNV at 0x7f2d113aa680>
adata.obs["cnv_prediction"].value_counts(dropna=False)
cnv_prediction
diploid 2561
aneuploid 412
NaN 27
Name: count, dtype: int64
Part.4 Genome-wide heatmap#
ov.pl.cnv_heatmap plots every cell as one row across the ordered
genomic bins. Gain = red, loss = blue, with an alternating chromosome
ideogram across the top. The groupby argument picks one
adata.obs column to control row ordering and the group-divider
lines. To compare cells under multiple labellings we draw two heatmaps —
the same X_cnv matrix re-sorted by cnv_prediction and by
cell_type — so we can read off whether the aneuploid band lines up
with the Epithelial-cell cluster. Rendering uses
marsilea for clean categorical
legends (pass backend='matplotlib' to fall back to the pure-matplotlib
renderer when marsilea isn’t installed).
# First view: ordered by the CopyKAT classification — aneuploid cells
# form a clean band at the top, diploid cells fill the rest.
ov.pl.cnv_heatmap(adata, groupby='cnv_prediction',
figsize=(8, 5), title='CopyKAT — ordered by cnv_prediction')
<marsilea.heatmap.Heatmap at 0x7f2d113aa470>

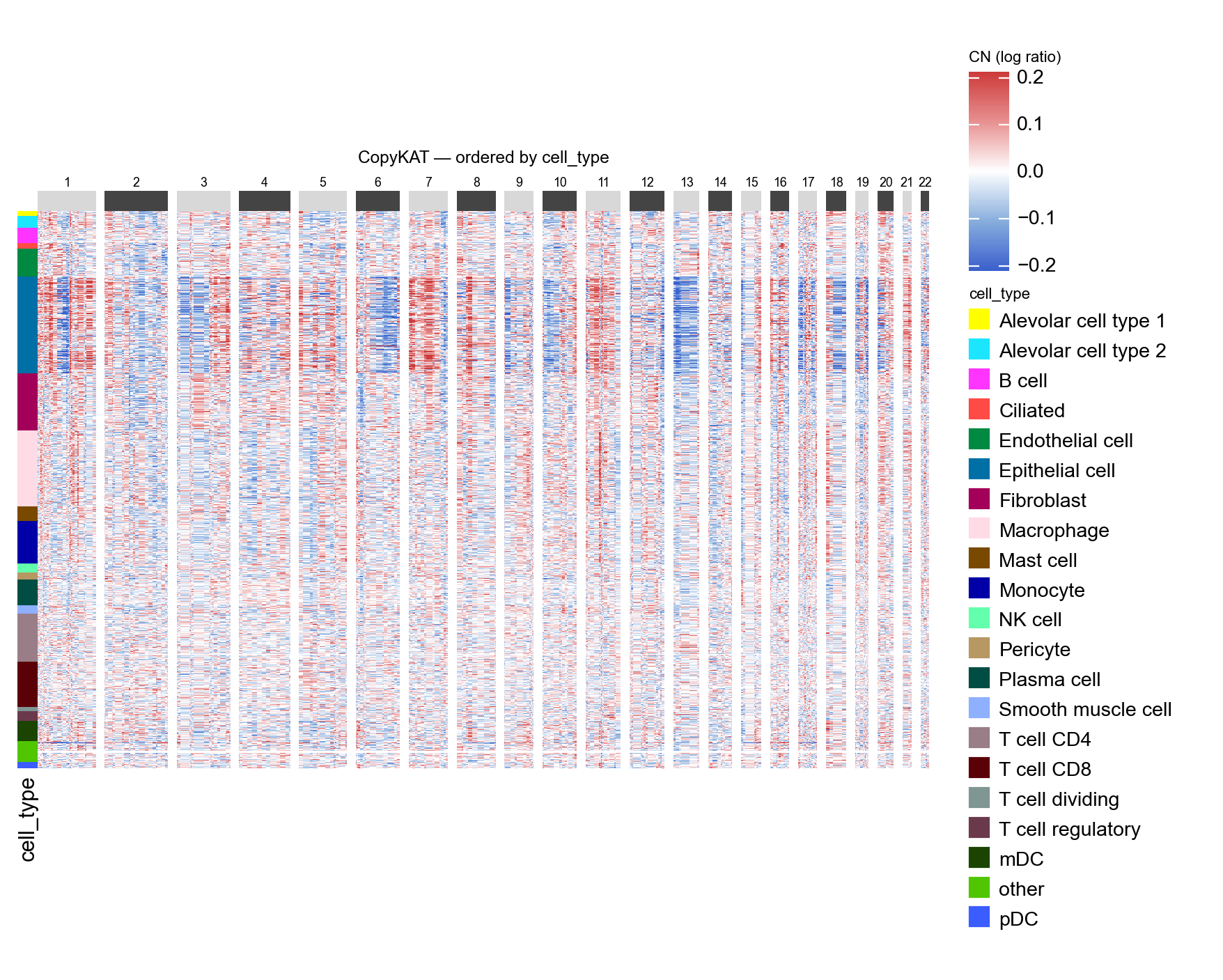
# Same matrix, re-ordered by cell_type. Recurrent CN patterns should
# concentrate inside the Epithelial-cell stripe.
ov.pl.cnv_heatmap(adata, groupby='cell_type',
figsize=(8, 5), title='CopyKAT — ordered by cell_type')
<marsilea.heatmap.Heatmap at 0x7f2f10384280>
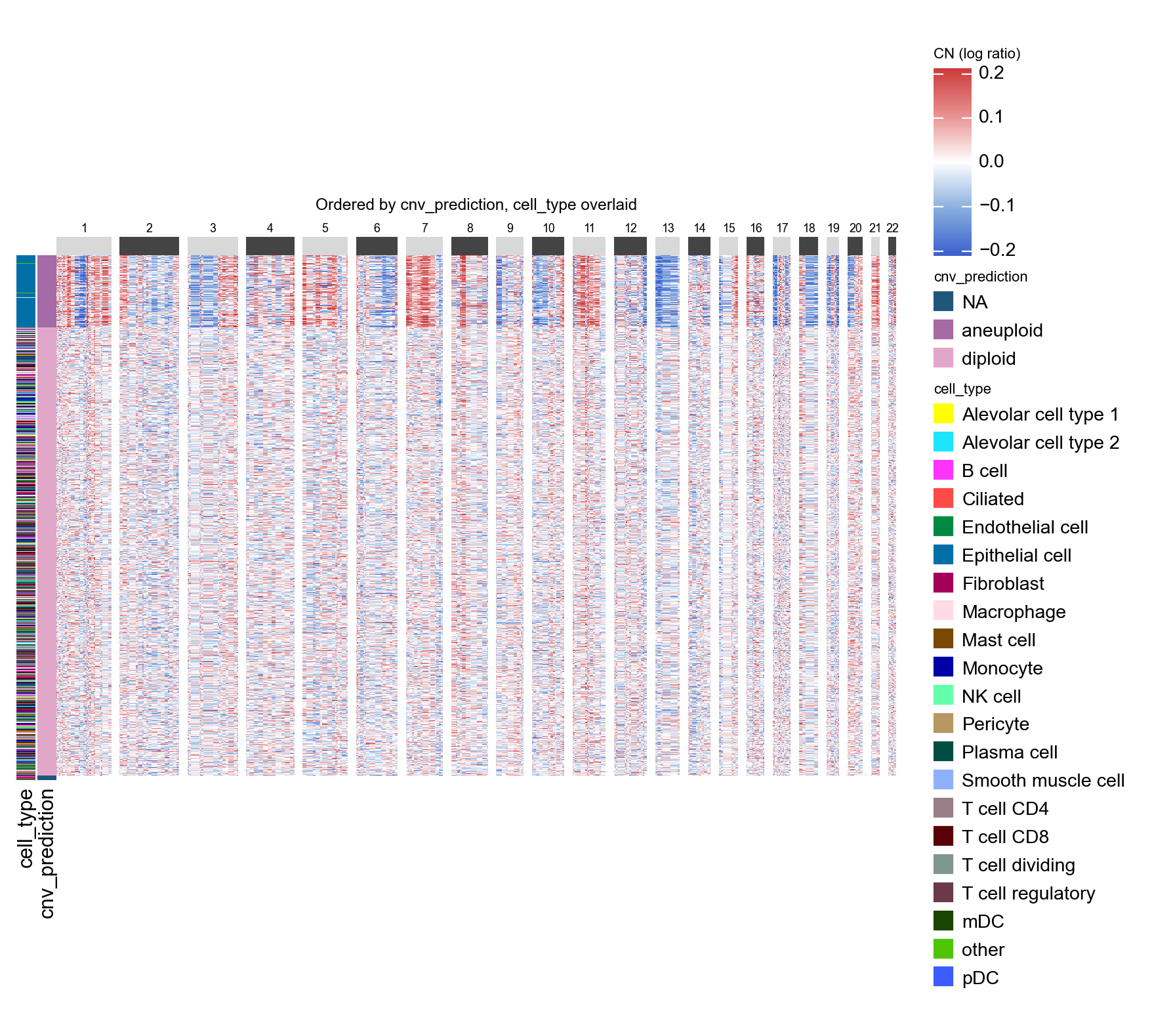
Layered annotation#
If you want the row ordering of one column with an extra colour strip
from another, pass the second column as annotations. The strip is
purely decorative — it does not re-order cells, so you can see at a
glance how a secondary category lines up against the primary grouping.
ov.pl.cnv_heatmap(adata, groupby='cnv_prediction', annotations=['cell_type'],
figsize=(8, 5),
title='Ordered by cnv_prediction, cell_type overlaid')
<marsilea.heatmap.Heatmap at 0x7f2ac7d222f0>
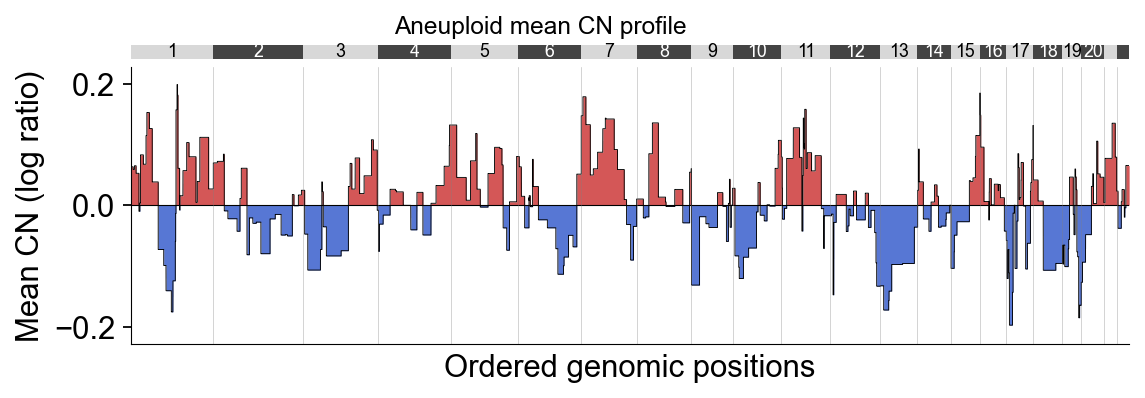
Part.5 Mean CN summary#
ov.pl.cnv_summary reduces the heatmap to a single line: the mean log-CN
across cells (or a subset) per genomic bin. Filling above zero
(gain_color) and below zero (loss_color) makes copy-number gain /
loss regions instantly visible. Below we plot the aneuploid mean — this
is the de-facto pseudo-bulk CNV profile of the tumour.
ov.pl.cnv_summary(adata, groupby='cnv_prediction', subset='aneuploid',
figsize=(8, 2.6), title='Aneuploid mean CN profile')
(<Figure size 640x208 with 2 Axes>,
{'line': <Axes: xlabel='Ordered genomic positions', ylabel='Mean CN (log ratio)'>,
'ideogram': <Axes: >})
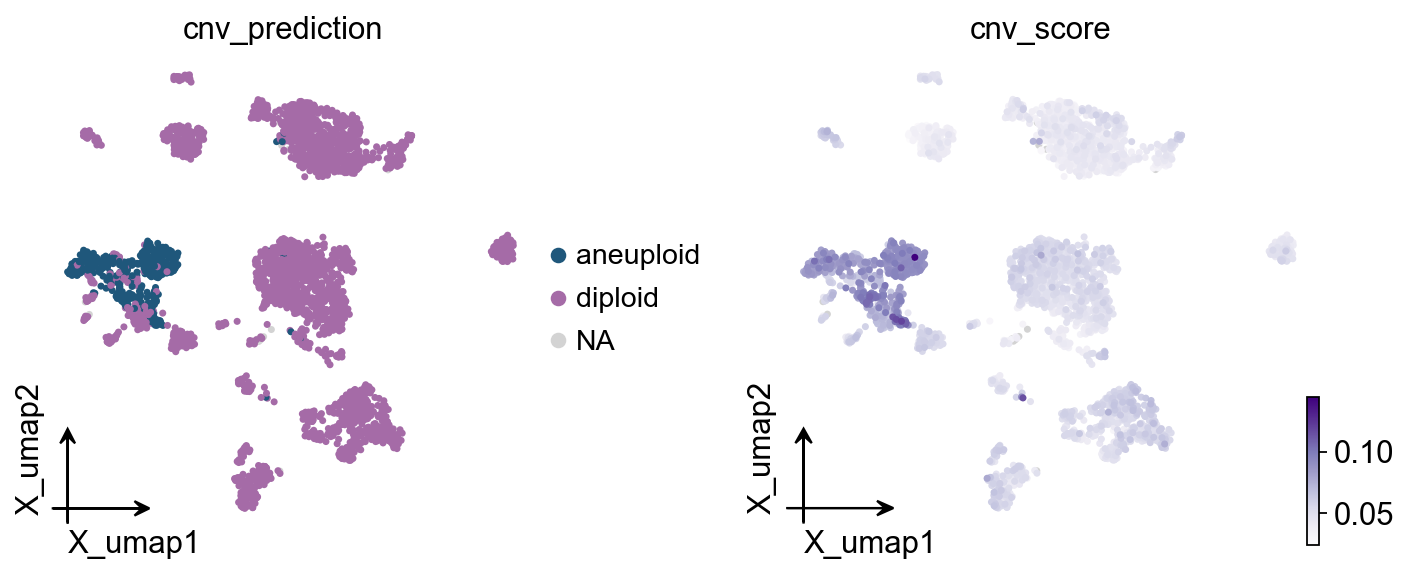
Part.6 UMAP#
The dataset already has a UMAP in adata.obsm['X_umap'].
ov.pl.cnv_umap colours it by the two columns CopyKAT writes —
cnv_prediction (categorical) and cnv_score (continuous). The
aneuploid cells should fall into one or a few coherent clusters that
overlap with the original Epithelial-cell annotation.
ov.pl.cnv_umap(adata)
Next steps: for reference-anchored CNV inference (when you have a
clean normal-cell annotation), see the
inferCNV tutorial. For an in-depth comparison
between CopyKAT and inferCNV on the same dataset, run both methods
end-to-end and visualise their cnv_score columns side-by-side with
ov.pl.embedding(adata, color=['cnv_score_copykat', 'cnv_score_infercnv']).