Multi-omics integration — metabolomics + RNA-seq with MOFA#
Most metabolomics studies that aim for mechanism also collect transcriptomics on the same samples. The standard unsupervised factor model for joint analysis is MOFA+ (Argelaguet 2020): it learns a small set of latent factors that capture variation shared across modalities plus modality-specific components.
omicverse ships MOFA+ via the vendored omicverse.external.mofapy2
plus the ov.single.pyMOFA trainer wrapper, but the downstream
helpers (pyMOFAART, factor_exact) are written for
single-cell AnnData where each row is a cell. For bulk
metabolomics studies each row is a sample, and those helpers are the
wrong fit.
ov.metabol.run_mofa is the metabolomics-friendly bridge: take a
dict of sample-aligned AnnData views, drive training, return the
factor × sample matrix.
This tutorial uses synthetic paired data (30 samples × 20 metabolites + 30 samples × 80 genes) with a planted shared factor so you can see MOFA+ recover it. For your real study you would pass your existing metabolomics AnnData together with your RNA-seq AnnData.
0 — Setup & simulated paired data#
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
from anndata import AnnData
import omicverse as ov
rng = np.random.default_rng(0)
n = 30
n_m = 20
n_g = 80
# Shared latent signal — some samples are high, others low
shared = rng.standard_normal(n)
# Metabolite view — first 8 metabolites load on the shared factor
X_m = rng.standard_normal((n, n_m)) * 0.3
X_m[:, :8] += 1.5 * shared[:, None]
# Gene view — first 30 genes load on the shared factor (higher-dim view)
X_g = rng.standard_normal((n, n_g)) * 0.3
X_g[:, :30] += 1.0 * shared[:, None]
samples = [f's{i:02d}' for i in range(n)]
obs = pd.DataFrame(index=samples)
adata_m = AnnData(X=X_m, obs=obs.copy(),
var=pd.DataFrame(index=[f'met{i}' for i in range(n_m)]))
adata_r = AnnData(X=X_g, obs=obs.copy(),
var=pd.DataFrame(index=[f'gene{i}' for i in range(n_g)]))
adata_m.shape, adata_r.shape
((30, 20), (30, 80))
1 — Train MOFA+#
Sample ordering must match across views — MOFA+ concatenates by
position, and run_mofa validates this and raises a helpful error
if it doesn’t. Training on 30 samples × 100 features takes <5 s.
factors = ov.metabol.run_mofa(
views={'metabol': adata_m, 'rna': adata_r},
n_factors=5,
outfile='mofa_demo.hdf5',
scale_views=True,
max_iter=200,
seed=0,
)
print(f'retained {factors.shape[1]} factor(s) after MOFA+ pruning')
factors.head()
#########################################################
### __ __ ____ ______ ###
### | \/ |/ __ \| ____/\ _ ###
### | \ / | | | | |__ / \ _| |_ ###
### | |\/| | | | | __/ /\ \_ _| ###
### | | | | |__| | | / ____ \|_| ###
### |_| |_|\____/|_|/_/ \_\ ###
### ###
#########################################################
Scaling views to unit variance...
Groups names not provided, using default naming convention:
- group1, group2, ..., groupG
Successfully loaded view='metabol' group='group0' with N=30 samples and D=20 features...
Successfully loaded view='rna' group='group0' with N=30 samples and D=80 features...
Model options:
- Automatic Relevance Determination prior on the factors: True
- Automatic Relevance Determination prior on the weights: True
- Spike-and-slab prior on the factors: False
- Spike-and-slab prior on the weights: True
Likelihoods:
- View 0 (metabol): gaussian
- View 1 (rna): gaussian
######################################
## Training the model with seed 0 ##
######################################
ELBO before training: -16211.58
Iteration 1: time=0.00, ELBO=-4208.27, deltaELBO=12003.310 (74.04156835%), Factors=4
Iteration 2: time=0.00, ELBO=-3502.03, deltaELBO=706.241 (4.35639954%), Factors=3
Iteration 3: time=0.00, ELBO=-3278.36, deltaELBO=223.667 (1.37967140%), Factors=2
Iteration 4: time=0.00, ELBO=-3086.21, deltaELBO=192.153 (1.18528216%), Factors=1
Iteration 5: time=0.00, ELBO=-3084.49, deltaELBO=1.724 (0.01063216%), Factors=1
Iteration 6: time=0.00, ELBO=-3083.47, deltaELBO=1.020 (0.00629021%), Factors=1
Iteration 7: time=0.00, ELBO=-3082.88, deltaELBO=0.590 (0.00364223%), Factors=1
Iteration 8: time=0.00, ELBO=-3082.54, deltaELBO=0.336 (0.00207426%), Factors=1
Iteration 9: time=0.00, ELBO=-3082.35, deltaELBO=0.190 (0.00116994%), Factors=1
Iteration 10: time=0.00, ELBO=-3082.25, deltaELBO=0.106 (0.00065633%), Factors=1
Iteration 11: time=0.00, ELBO=-3082.19, deltaELBO=0.060 (0.00036709%), Factors=1
Iteration 12: time=0.00, ELBO=-3082.15, deltaELBO=0.033 (0.00020498%), Factors=1
Converged!
#######################
## Training finished ##
#######################
Saving model in /scratch/users/steorra/analysis/omicverse_dev/omicverse/omicverse_guide/docs/Tutorials-metabol/mofa_demo.hdf5...
retained 1 factor(s) after MOFA+ pruning
F1
s00 -0.694762
s01 0.088217
s02 -2.239374
s03 -0.595385
s04 1.260085
2 — Plug the factors back into an AnnData#
The returned DataFrame is samples × factors. The standard move is
to attach it as obsm['X_mofa'] so the rest of omicverse (plotting,
clustering) can see the integrated embedding.
adata_m.obsm['X_mofa'] = factors.reindex(adata_m.obs_names).to_numpy()
adata_r.obsm['X_mofa'] = factors.reindex(adata_r.obs_names).to_numpy()
print('metabol AnnData now carries:', list(adata_m.obsm.keys()))
metabol AnnData now carries: ['X_mofa']
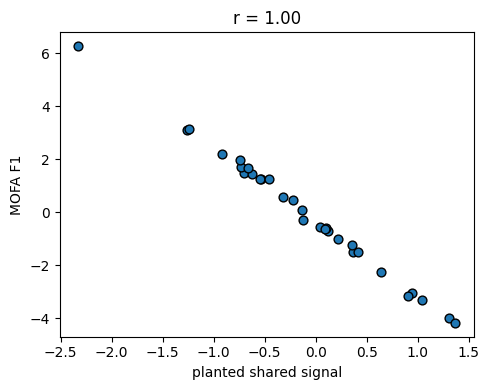
3 — Recovery of the planted shared factor#
Correlate each MOFA factor against the shared signal we planted. At least one factor should track it closely.
corrs = factors.corrwith(pd.Series(shared, index=samples)).abs()
print('Factor correlation with planted shared signal:')
print(corrs.sort_values(ascending=False))
best = corrs.idxmax()
fig, ax = plt.subplots(figsize=(5, 4))
ax.scatter(shared, factors[best], color='C0', edgecolor='k', s=40)
ax.set_xlabel('planted shared signal')
ax.set_ylabel(f'MOFA {best}')
ax.set_title(f'r = {corrs[best]:.2f}')
fig.tight_layout()
plt.show()
Factor correlation with planted shared signal:
F1 0.998612
dtype: float64
4 — How to use factors downstream#
Clustering / sub-typing: apply k-means or hierarchical clustering on
factors.Association testing: correlate each factor against a clinical variable (age, response, dose).
Factor interpretation: load the HDF5 at
factors.attrs['hdf5_path']withmofaxto inspect per-view loadings; metabolites or genes with high absolute loadings on factor X drive that factor.
Notes on real data#
Normalize each view first (metabol: PQN + log; RNA-seq: log-CPM or log-normalized raw counts).
Scale views (
scale_views=True, the default) so a higher-dim view doesn’t dominate.Check the residual variance per view in the HDF5; low shared factor share → the two modalities don’t actually co-vary, and MOFA+ is the wrong tool.